

Abstract and Introduction
Abstract
Objective
Germline mutations in the tumor suppressor genes BRCA1 and BRCA2 predispose women to breast and ovarian cancer. Female carriers of BRCA1 or BRCA2 gene mutations have very high lifetime risks for breast and ovarian cancers. Genetic abnormalities occur in all cancers, so BRCA-related pathways are critical because they serve to safeguard genetic content. Although protecting genetic information is a general function, BRCA-related pathways seem largely specific to preventing breast and ovarian cancer. The objective of this study was to resolve this difference between the theoretical functions of BRCA genes and their specific clinical effects.
Data Sources, Data Extraction, Data Synthesis
The author collected data published in > 30 epidemiologic studies on the incidence of cancers other than breast or ovarian in mutation carriers and in large populations eligible for mutation testing. Data were extracted and used directly as published whenever possible with a minimum of statistical manipulation.
Conclusions
Although mutations target breast and ovary, a broader spectrum of cancers also occur with statistically significant elevated frequencies. Risks for “all cancers except breast or ovary” are elevated, with some population subgroups differing with regard to how frequently elevated risks were found at individual sites. Additional sites at risk included stomach, pancreas, prostate, and colon. The increased risk ranged from about 20% to 60%, with the greatest increases in risk in stomach and pancreas. The collected data show BRCA-pathway functions are probably required at multiple sites, not just in breast or ovary. Known interactions and relationships among BRCA-related pathways strongly support the idea that their inactivation provides growth or survival advantages for a variety of cancers. The data suggest applying an increased level of clinical alertness to those with defects in BRCA-related pathways. Identifying molecules that confer growth or survival advantages to BRCA-related cancers may provide broadly useful targets for chemotherapy or chemoprevention.
Introduction
Germline mutations in the tumor suppressor genes BRCA1 and BRCA2 predispose women to breast and ovarian cancer. Some female carriers of inherited mutations in BRCA1 or BRCA2 genes have lifetime risks for breast cancer exceeding 80%.
BRCA-related pathways safeguard genetic content, and a very large fraction of human cancers have abnormal genetic content. BRCA1 and/or BRCA2 are involved in pathways important for DNA damage recognition, double-strand break repair, checkpoint control, transcription regulation, and chromatin remodeling. These functions are essential and important for all cell types. Despite the general nature of BRCA functions, tumors in mutation carriers predominantly target breast and ovary. A major problem is that the broad theoretical importance of BRCA-related pathways conflicts with the specific targeting of cancers to breast and ovary. Some observational studies report elevations of the risk for certain cancers besides breast or ovarian cancer in BRCA1 or BRCA2 mutation carriers, but other studies conflict.
Although mutations in BRCA-related pathways may increase risk for sporadic or hereditary cancers other than breast or ovarian, the risks for these additional cancers may be smaller than those for breast or ovarian cancer; they may show wider variation; they may require large populations to measure; and they may involve proteins elsewhere in the pathways.
Carriers of mutations in BRCA1 and BRCA2 are relatively infrequent in the general population, and BRCA mutations are thought to be involved in at most 5% to 10% of all breast cancers. Mutations of genes encoding ATM and CHEK2 or mutations leading to EMSY amplification also affect BRCA pathways. These changes occur more frequently than BRCA1 or BRCA2 mutations in the general population, suggesting inactivation of BRCA-related pathways is probably associated with significantly higher percentages of cancers. Fortunately, data exist from many thousands of high-risk individuals who were not identified mutation carriers but who were eligible for mutation testing. These data would include all mutations that affect BRCA pathways, and some studies provide a built-in control because they also include those who were not eligible for mutation testing.
I collected data published in more than 30 epidemiologic studies on the incidence of cancers other than breast or ovarian in mutation carriers or in large populations eligible for mutation testing. Results of meta-analyses show that the loss of BRCA gene function provides growth or survival advantages to a broader spectrum of tumors, including stomach, pancreas, prostate, and colon.
Thus, BRCA pathways function at multiple sites throughout the body, not just in breast or ovary. This does much to resolve a major conflict between the mechanisms of BRCA pathways participation in tumor suppression and clinical observations. The data suggest there ought to be increased clinical awareness for those with defects in BRCA-related pathways. Identifying molecules that confer growth or survival advantages to tumors in those with BRCA pathway deficits may provide helpful targets for chemotherapy or chemoprevention.
Methods
Search Strategies
Many PubMed searches were conducted. To search for possible effects between BRCA mutations and sporadic cancers, individual cancers were entered so the search read “BRCA1 and melanoma” and then “BRCA2 and melanoma.” Initially, the term “colon cancer and BRCA” was entered. Then all possible variants were entered such as “colorectal cancer and BRCA1.” An additional strategy was “hand searching” by consulting appropriate references in literature that seemed pertinent. A few searches of EMBASE were also done. Additional searches were conducted for incidence of cancers in high-risk groups, for second cancers after diagnosis of primary breast or ovarian cancer at young ages, and for second cancers after diagnosis of male breast cancer.
Studies Included
More than 30 observational studies going back 20 years and including case histories from many thousands of mutation carriers or possible mutation carriers in high-risk populations were collected and intensively reviewed. Collection of data representing a large number of mutation carriers was achieved by including several types of studies as follows. Population studies were used that tabulated cancer histories for known mutation carriers and for their first-degree relatives. Large populations enriched in mutation carriers, such as those eligible for BRCA gene mutation testing, were also included. A third type of study measured incidences of second cancers after breast cancer in young women. The likely number of BRCA mutations was then estimated from data in the publication or from mutation incidence tables available at www.myriadgenetics.com. I also included studies that measured whether the frequency of BRCA gene mutations was elevated in patients presenting with certain cancers other than breast or ovarian.
Statistics
The literature results were used directly as reported by the original authors. This enabled inclusion of data that were 10 to 20 years old in addition to the most recent studies. The goals for data analysis were to use as little additional statistical manipulation as possible such that it would allow anyone to reproduce the results. The summary method of meta-analysis using the StatsDirect Computer Program was used for statistical calculations. StatsDirect calculates stratum weights as the inverse of the variance for the summary statistic (Y) supplied. The pooled estimate of Y is calculated as a weighted mean, ie, the sum of weighted Y for each stratum divided by the sum of the weights. Meta-analyses based on approximate relative risk calculations were also run as a double-check using available data in the original publications.
Exclusion Criteria
Studies were excluded in whole or in part if they did not present relative risks and confidence intervals or provide enough data to calculate missing information. Because of the close interactions of Fallopian tubes and ovaries and because data show that Fallopian tubes are highly susceptible in BRCA mutation carriers, Fallopian tube cancer was considered to be a part of the breast/ovarian cancer syndrome.
Format
Data from the studies included sample size, population, method, control population, method of expressing results (as relative risks, percentages, odds ratios, etc), and 95% confidence interval.
Reliability of Diagnoses
I found no evidence for misdiagnosis of cancer, and most studies reported histologic verification in a high percentage of cases. It was necessary to make the assumption that cancers were diagnosed correctly.
Results
BRCA Mutations and Sporadic Cancers: Primary Observational Data
Studies used are numbered and summarized in Table 1 and Table 2. Table 1 contains population studies (numbered 1-17) that include data on the incidence of cancers other than breast or ovarian cancer in BRCA mutation carriers or populations likely to be enriched in mutation carriers. Table 2 reports the measured incidence of BRCA1 or BRCA2 mutations in families of patients presenting with a particular cancer.[1–33]
Table 1.
Epidemiologic Studies That Include Data on the Risk of “Other” Cancers
| Reference Authors, Year | Study Group/characteristics | Comparison Group | Number of mutation carriers or likely mutation carriers in study | Cancers other than breast, or ovarian in carriers or relatives where p≤.05 and/or confidence interval does not include 1. RR= relative risk. SIR= Standardized Incidence Ratio. (95% confidence interval in brackets.) | ||
|---|---|---|---|---|---|---|
| 1. Ford et al 1994 | 464 BRCA1 carriers and their relatives from 29 families in North America and 4 in England and Wales. | Rates from SEER program of NCI used to determine expected rates. England & Wales rates for 4 families. | 464 BRCA1 mutation carriers | RR [95%CI] | ||
| Colon | 4.11 [2.36–7.15] | |||||
| Prostate | 3.33 [1.78–6.20] | |||||
| All cancers except breast ovary and non-melanoma skin | RR=1.44 | |||||
| 2. Breast Cancer Linkage Consortium 1999 | 173 families with BRCA2 mutations identified at 20 centers in Western Europe and N. America. All pathologic BRCA2 mutations included. Assumes uniform risk across all mutations | From publication “Cancer Incidence on five continents”, SEER data for US from 1973 on. | 3728 individuals., including 50 men with breast cancer, 631 women with breast cancer below age 60 or ovarian cancer at any age. | Data for all ages from 0–85: | RR [95%CI] | |
| Buccal cavity and pharynx | 2.26 [1.09–4.68] p=.06 | |||||
| Stomach | 2.59 [1.46–4.61] | |||||
| Pancreas | 3.51 [1.87–6.58] | |||||
| Gallbladder/bile duct | 4.97 [1.50–16.52] | |||||
| Malignant melanoma | 2.58 [1.28–5.97] | |||||
| Prostate | 4.65 [3.48–6.22] | |||||
| All cancers except breast and ovary | 1.90 [1.63–2.23] | |||||
| All cancers except breast, ovary, prostate and pancreas | 1.47 [1.21–1.79] | |||||
| Other or ill-defined sites | 4.13 [2.05–8.32] | |||||
| Liver cancer? | 4.18 [1.56–11.23] p >0.05 | |||||
| 3. Thompson et al 2002 | Cancer risks in BRCA1 carriers 2245 tested positive from 30 centers in Western Europe and North America. Largest study to date had statistical power to detect more moderate risks. | Taken from the publication “Cancer incidence on five continents” averaged over all countries represented in the study. | 2245 | Colon | 2.03 [1.45–2.85] | |
| Liver | 4.06 [1.77–9.34] | |||||
| Pancreas | 2.26 [1.26–4.06] | |||||
| Uterine body | 2.65 [1.69–4.16] | |||||
| Cervix | 3.72 [2.26–6.10] | |||||
| All except breast, ovary, and non-melanoma skin | 1.34 [1.19–1.51] | |||||
| Intestinal tract and other sites | 7.40 [5.14–10.66] | |||||
| Unknown site | 3.45 [2.35–5.07] | |||||
| 4. Brose et al 2002 | Clinic based study analyzing a subset of patients making up about 15% in study 3. 381 females from 147 families with documented BRCA1 mutations. | SEER 2000 data | 381 | Colon | 2.0 [1.5–2.5] | |
| Gastric | 6.9 [4.25–9.38] | |||||
| Pancreatic | 2.8 [1.46–4.07] | |||||
| No elevation found for prostate cancer | ||||||
| 5. Johannsson et al 1999 | Cancer incidence between 1958 and 1995 from data in Swedish cancer registry, cause of death registry and census registry. | Age gender and calendar year specific reference data from all of Sweden. | 1873 individuals related to 29 BRCA1 probands and 20 BRCA2 probands. Mutation testing was done only for probands. | Women in BRCA1 families: | Standardized mortality ratios (SMR) | |
| Stomach cancer | 5.86 [1.60–15.01] | |||||
| Men in BRCA1 families: Squamous cell skin cancer | 6.02 [1.96–14.05] | |||||
| BRCA2 families including index cases: | ||||||
| invasive cervical cancer | 4.21 [1.15–10.8] p=0.016. | |||||
| Prostate | borderline significance | |||||
| pancreatic | Not increased | |||||
| 6. Evans et al 2001 | Women diagnosed with breast cancer at age<50 in SE England. Identified multiple primary additional cancers via the Thames cancer database. | Cancer rates observed in the corresponding region during the same time period. | 32,799 women diagnosed with breast cancer at age<50 predicting at least 1476 mutation carriers. | Esophagus | 2.39 [1.6–3.57] | |
| Stomach | 1.83 [1.29–2.59] | |||||
| Myeloid leukemia | 2.31 [1.52–3.01] | |||||
| Lung, bronchus | 1.49 [1.26–1.78] | |||||
| All sites excluding breast | 1.21 [1.13–1.31] | |||||
| Uncertain (CI includes 1): | ||||||
| Pancreas | 1.34 [0.9–1.99] | |||||
| Thyroid | 1.74 [0.99–3.07] | |||||
| 7. Harvey and Brinton 1985 | Primary cancers after diagnosis of initial breast cancer in females younger than 45 in Connecticut | Expected rates given for group but methods not explained. | About 7.3–12.2% based on Myriad tables, depending on percentage of Ashkenazi Jewish people. | Second site | RR [CI calculated] | |
| Colon+rectum | 1.68 [1.12–2.52] | |||||
| All excluding breast cancer | 1.44[1.19–1.75] | |||||
| 8. Teppo et al 1985 | New primary cancers after diagnosis of the first primary cancer. Data from Finnish Cancer registry. | About 5297 cancers were calculated to occur to women younger than 50. | Only lung cancer was significantly elevated RR=3.11[1.2–8.08] | |||
| But in aggregate any cancer excluding breast and ovary had RR=1.42[1.10–1.83] | ||||||
| 9. Bermejo and Hemminki 2004 | Data in Swedish cancer registry including at least 3 generations. Families eligible for BRCA1/2 test. Mutation frequencies in Germany were used. | Compared incidences in general population. | Estimated at 20984 based on percentages of mutation carriers in subgroups in the publication. Myriad Genetics Tables were used if percentages were not given. Group with 2 breast cancers after 50 was excluded because of low odds of mutation carriers. | Cancer | Significant SIR | Subgroup |
| Pancreas before 50 | 5.5 [1.43–14.2] | 2 bcs < 50 | ||||
| Liver primary | 1.77 [1.01–2.88] | 2 bcs one < 50 | ||||
| Prostate | 1.18 [1.01–1.37] | 2 bcs one < 50 | ||||
| Prostate | 1.31 [1.05–1.62] | 2 bcs < 50 | ||||
| Prostate | 1.45 [1.11–1.87] | bilateral bc < 50 | ||||
| Prostate before 65 | 1.58 [1.14–2.13] | 2 bcs one < 50 | ||||
| Pancreas | 1.48 [1.05–2.04] | 1 bc < 35 | ||||
| Pancreas before 50 | 6.54 [1.23–19.4] | bilateral bc < 50 | ||||
| Pancreas before 50 | 5.50 [1.43–14.2] | 2 bcs < 50 | ||||
| Pancreas | 1.91 [1.04–3.22] | bilateral bc < 50 | ||||
| Eye | 3.84 [1.00+-9.92] | bc and oc | ||||
| Stomach before 70 | 2.04 [1.14–3.12] | bc and oc | ||||
| 10. Shih H, et al. 2000 | 98 women in UMichigan or UPenn clinics with breast cancer reporting at least 1 other primary cancer in themselves or in a relative with breast cancer. | 99 women with only breast cancer. | 98 | 42.9% of families reporting breast and any second nonbreast primary cancer have a BRCA1/2 mutation vs 12.1% of those with breast cancer only. (p<0.0001.) Odds ratio associated with carrying either a BRCA1 or a BRCA2 mutation in the nonovarian multiple cancer cases vs families reporting only breast cancer was 2.13 [0.93-4.90] Primary cancers included colorectal, cervical, endometrial, thryroid, leukemia, and lymphomas. Increased risk for prostate and pancreatic cancer not reported | ||
| 11. Easton et al 1997 | Cancer risks in 2 large breast cancer familes linked to BRCA2 | SEER rates or published rates from England and Wales if appropriate. | Extended families including nearly 500 individuals. | Laryngeal cancer RR7.67 (p<.01) based on 2 cancers. Prostate RR=2.89 (p<.01). Confidence intervals were not given. | ||
| Ocular melanoma was also found. Authors pointed out the need for larger studies and additional data. | ||||||
| 12. Moslehi et al 2000 | Calculated relative risks for cancers in first degree relatives of Ashkenazi Jewish ovarian cancer patients vs controls. | 368 female relatives of noncarriers and 349 male relatives of non carriers (Table 6 of publication) | 253 relatives of female probands and 223 male relatives of 86 probands. | Relative risk | ||
| Cancer to age 75 excluding breast and ovarian cancer | 1.5 [1.0–2.1] | |||||
| Endometrial cancer to age 75 in relatives of BRCA1 carriers | 9.4 p=0.0003 | |||||
| Significant increase in prostate cancer risk for carriers of BRCA1 (p=0.01) or BRCA2 (p=0.002) | ||||||
| 13. Risch et al 2001 | 649 unselected incident cases of ovarian cancer diagnosed in Ontario, Canda from 1995-96 tested for BRCA mutations. | 4378 relatives of cases who did not carry a mutation. | 60 mutations identified included 39 BRCA1 mutations and 21 BRCA2 mutations. | RR to age 80 in 1st degree relatives of BRCA1 mutation carriers: | ||
| Stomach | 6.2[2–19]; | |||||
| Leukemias | 2.6[1.02–6.6] | |||||
| Relatives of BRCA2 mutation carriers | ||||||
| Colon rectum | RR=2.5[1.02–6.3] | |||||
| Relatives of those with BRCA2 Mutations in ovarian cancer cluster | ||||||
| Ovarian, colorectal, stomach, pancreatic, OR prostate cancer | 3.1 [1.7–5.7] p=0.0003 | |||||
| 14. Aretini et al 2003 | Families with BRCA1/2 mutations ascertained in 6 italian centers | Not explained. | 179 proband mutation carriers and 66 mutation carriers among relatives. | 440 1st or 2nd degree relatives affected by breast and/or ovarian cancer. 230 other cancers excluding breast, and ovarian; 200 if prostate and pancreatic are also excluded. Cancers in BRCA1 families included lung, gastric uterine, colon rectum, hepatobiliary, CNS, GI, and others. These occurred in significant numbers of families but risk and incidence ratios were not calculated. Pancreatic cancer RR=2.03; Prostate cancer RR=1.91 | ||
| 15. Streuwing et al 1997 | 5318 Jewish people over age 20 in Washington DC area. Compared cancer histories in 306 female and 273 male first degree relatives of mutation carriers | 13,018 female and 13,324 male first degree relatives non-carriers. | 120 (61 with BRCA1 founder mutations and 59 with BRCA2 founder mutation) | Increased % of lung pancreatic, lymphoma, uterine cancer, multiple myeloma, thyroid cancer, Hodgkin's disease, and stomach cancer but the results were based on low numbers of carriers. Reduced incidence of colon cancer among carriers. 16% risk of prostate cancer at age 70 [4–30%] for BRCA1 or BRCA2 mutations. Observed elevation in pancreatic cancer was not statistically significant. | ||
| 16. Goldgar et al 1994 | Estimated familial risks in Utah population database (∼250,000) records by identifying all cases of cancer in first degree relatives. | Cohort-specific internal rates calculated from 399,786 relatives of all individuals in Utah database who died in Utah. | 1145 first degree relatives of women had breast cancer that developed at <50 years. (7.3% mutation carriers predicts ∼83.5 women) | Second site | RR [CI] | |
| Colon | 1.72 [1.3–2.2] | |||||
| Non-Hodgkins lymphoma | 1.92 [1.3–2.7] | |||||
| Prostate | 1.36 [1.1–1.7] | |||||
| 17. Berman et al 1996 | 83 Ashkenazi jewish individuals with cancer and 93 diagnosed with ovarian cancer at any age. | Pedigree study only. | 8 carriers of the BRCA2 mutation 6174delT. | Several of the mutation carriers had significant cancer histories besides breast or ovarian cancer. These histories included an increased incidence of colon, esophageal, pancreatic stomach and hematopoietic cancers. Lung, brain, and endometrial tumors were also observed. | ||
*P ≤ .05 and/or confidence interval does not include 1.
RR = relative risk; SIR = standardized incidence ratio (95% confidence interval in brackets)
Table 2.
Studies of Patients With Individual Cancers Other Than Breast or Ovarian Tested for Association With BRCA Mutations
| Reference | Study Group | Comparison Group | Number of Mutation Carriers or Likely Mutation Carriers in Study | Risks for Cancers Other Than Breast, Ovarian, Prostate, or Pancreas* | Cancer Rrisk in Carriers or Incidence of Carriers in Cancer Patients |
|---|---|---|---|---|---|
| 18. Hahn et al, 2003 | Identified 26 European families in which at least two first-degree relatives had a histologically confirmed diagnosis of pancreatic ductal adenocarcinoma. Typed members of families for BRCA2 mutation | 64 | The median age at diagnosis was 60 yrs (range 33–81 yrs); 14 (22%) patients were younger than 50 yrs at diagnosis. 5 families had at least one member diagnosed with breast cancer; 3 families had at least one member diagnosed with colon cancer; 2 families had at least one member diagnosed with prostate, gastric, or lung cancer; and 4 families had one member diagnosed with either head and neck, esophageal, or ovarian cancer or osteosarcoma. | RR pancreatic cancer = 3.5. Prostate cancer was also found but risk was not calculated. | |
| 19. Murphy et al, 2003 | Analyzed 31 samples of DNA from cancer patients in pancreatic cancer kindreds with 3 cases and at least 2 affected people first-degree relatives. 29 kindreds, 6 Ashkenazi Jewish, 10 not-Jewish and 13 unspecified | Used national familial pancreas tumor registry to compare ages of onset | 5 mutation carriers. One patient had a point mutation of uncertain significance | Melanoma, and cancers of liver, brain, skin, colon, lung, brain, prostate, testes, and bladder were also found in the kindred. Only 1 patient had a family history of breast cancer and no ovarian cancer. No difference in age of onset in comparison | 17% of patients from familial pancreatic cancer kindreds had pathogenic BRCA2 mutations. 1 case of prostate cancer noted in the kindred. |
| 20. Lal et al, 2000 | 102 study patients with pancreatic adenocarcinoma. Family history by questionnaire. BRCA mutations in screened by PTT | 11 patients with early onset disease or multiple primary cancers without a family history | 4 | Most high and intermediate risk families did not have detectable germ-line mutations. 4% of patients had histories suggestive of BRCA1, or BRCA2 mutations. Numerous other cancers were found in BRCA1 and BRCA2 mutation carriers including lung cancer, leukemia, brain cancer, skin cancer, lymphoma, and colon cancer. | 31% of Ashkenazi Jewish patients were BRCA2 mutation carriers. |
| 21. Kirchhoff et al, 2004 | Case-control study that screened blood from 251 Ashkenazi men with prostate cancer | 1472 male Ashkenazi volunteers without prostate cancer were controls | 13 | OR for BRCA2 mutation carriers = 4.78 [1.87–12.25] BRCA1 carriers were not significantly affected. | Prostate cancer OR = 4.78 [1.87–12.25] |
| 22. Edward et al, 2003 | Screened 263 men with prostate cancer in UK, who were younger than 55 for mutations in BRCA2 coding sequence. | Estimates from previous studies of general population. | 6 | Relative risk of developing prostate cancer by age 56 was increased 23-fold. | Prostate cancer before age 56: 23-fold increased risk |
| 23. Vazina et al, 2000 | Tested 174 unselected Jewish Israeli prostate cancer patients for BRCA1/2 founder mutations | Used historical controls as in Struewing 1997 | 5 | Rates of predominant BRCA1 mutations in prostate cancer patients (2.3% and 3.3%) was greater than the rate of the general population (0.77% and 0.37%). Mutation carriers had double the risk of the general Ashkenazi population. Only 1 of 5 carriers had prostate cancer detected early. | See column 5. Pancreatic cancer not reported. |
| 24. Hubert et al, 1999 | Unselected 87 Ashkenazi Jewish prostate cancer patients | Israeli data for rate of carriers | 3 | Expected 11 cases of prostate cancer in BRCA1/BRCA2 mutations but observed only 3 | Prostate cancer risk not elevated |
| 25. Thorlacius et al, 1998 | 34 men | Families negative for the mutation | 13 | Risk for carriers was higher than for noncarriers but the result was marginally significant at age 63. | Marginal |
| 26. Sigurdsson et al, 1997 | Prostate cancer cases from 16 BRCA2 families and all available samples from Iceland prostate cancer patients | Families negative for the mutation | 8 | Risk ratio of prostate cancer was 4.6 [1.9–8.8] in first-degree relatives and 2.5 [1.2–4.6] in second-degree relatives of 16 BRCA2 probands. BRCA2 mutation may be a marker for an aggressive form of prostate cancer. | See column 5. Pancreatic cancer not reported. |
| 27. Iscovich J et al, 2002 | Examined 143 Israeli cases of uveal melanoma for the presence of the 6174 delT BRCA2 mutation. | Used published mutation frequencies for particular BRCA2 mutation. | 4 | Mean age of uveal melanoma in mutation carriers was 47.5 vs. 60.6 in non-mutation carriers. | 2.8% of patients had BRCA2 mutation [0–5.6] |
| 28. Scott et al, 2002 | Investigated BRCA2 mutations in 69 ocular melanoma patients age <50 in Australia | 6 | 3% prevalence of ocular melanoma in patients younger than 50 | Not reported | |
| 29. Neill et al, 2004 | Population-based case-control study in northern Israel. 1422 case patients with incident colorectal cancer, diagnosed between March 31, 1998, and December 31, 2002. Genotyped for 3 BRCA1/2 founder mutations. | 1566 control subjects without colorectal cancer from northern Israel | 24 | Cumulative cancer risk for any cancer type except breast or ovarian cancer was statistically significantly greater in first-degree relatives of BRCA1/2 mutation carriers (by age 50 years: 3.6%, [0.4% to 6.7%]; by age 70 years: 13.7%, [6.0% to 21.5%] compared with first-degree relatives of BRCA1/2 mutation non-carriers (by 50 yrs: 1.7%, [1.4% to 2.0%]; by 70 yrs: 8.0%, [7.1% to 8.9%]. | Modest or no elevation in colon cancer risk with BRCA2 mutation |
| 30. Kirchhoff et al, 2004 | Screened for 3 BRCA founder mutations 586 unselected Ashkenazi Jewish colorectal cancer patients. Adjusted for age and sex with logistic regression analysis. | 5012 Ashkenazi Jewish patients without a known history of colorectal cancer | 6 | BRCA mutation status was not statistically significantly associated with colorectal cancer. | Not reported |
| 31. Chen-Stoyerman et al, 2001 | Analyzed peripheral blood lymphocytes of Ashkenazi Jewish colorectal cancer patients for 3 founder mutations. 225 consecutive colorectal cancer patients. Ashkenazi origin confirmed for 3 generations. 125 men and 100 women | Published data for the general Ashkenazi Jewish population | 4 | Frequency of colorectal cancer was similar to the frequency in the general Ashkenazi population. | Not reported |
| 32. Drucker et al, 2000 | 136 consecutive Israeli Jewish patients with colorectal cancer were screened for 3 founder mutations | 3 | Preliminary study 3 carriers out of 87 tested for BRCA mutations suggested mutation carriers were at increased risk for colon cancer. | Not reported |
Relative Risk (RR), Odds Ratio (OR), or standardized incidence ratio (SIR) (95% confidence interval in brackets)
Table 1 lists cancers that occurred at a statistically significant greater frequency relative to controls if BRCA function is impaired in at least some subgroups of mutation carriers within individual studies (column 6): colon cancer; colorectal cancer; gall bladder/bile duct cancer; liver cancer; stomach cancer; malignant melanoma; esophageal cancer; myeloid leukemia; lung/bronchus cancer, laryngeal cancer, Hodgkin's disease, cancer of the uterine body and cervix; testicular cancer; cancer at multiple sites, including pituitary and peritoneum; all cancers except breast ovary, nonmelanoma skin; all cancers in females other than breast, ovarian and nonmelanoma skin; any second non-breast primary cancer; and unknown primary site cancer.
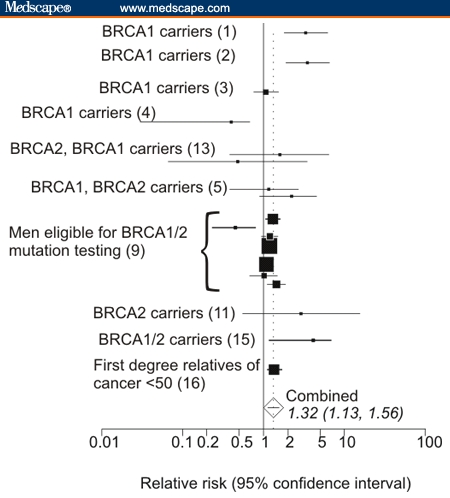
Figure 1A plots the statistically significant relative risks extracted from the 17 studies cited in Table 1. The plot shows that the increased incidences of cancers are not consistent among studies and vary in studies of different population subgroups. For example, 6 of 17 studies show statistically significant elevated risks for stomach cancers in some subgroups, and 4 studies show elevated risk for pancreatic cancer. Even different groups within the same study show variation. For example, comparisons of pancreatic cancer risks among different individual groups in the study of Bermejo and Hemminki[9] show wide variation (Table 1), but their aggregated data include 1 as part of the confidence interval. Increased risks for esophageal, lung, non-Hodgkin's lymphoma (NHL), malignant melanoma, buccal cavity/pharynx, squamous skin, and gallbladder/bile duct cancer are each represented in only 1 study. (Gallbladder/bile duct cancer was plotted near the results for liver cancer in Figure 1A).
Figure 1.
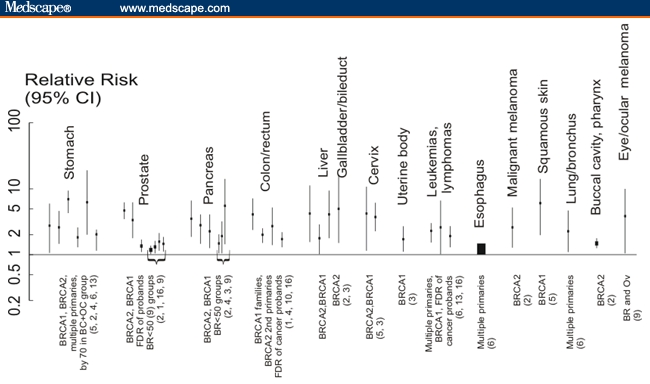
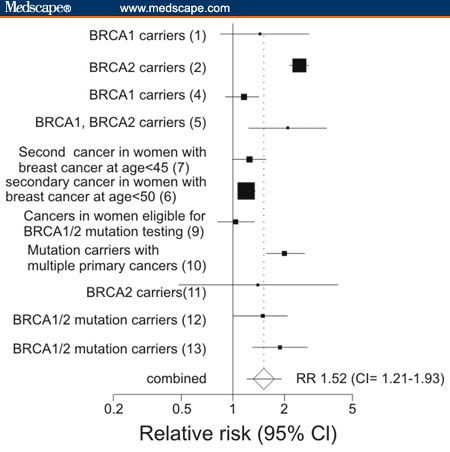
A. Significant relative risks for cancers other than breast/ovarian in mutation carriers or likely mutation carriers found in various subgroups by different studies. Sites are arranged in descending frequency that statistically significant cancers were reported in different publications. Brackets indicate reports from the same publication for different groups. Gallbladder/bile duct cancers are grouped together as are leukemias and lymphomas. Population groups are listed along the bottom of the graph with references in parentheses. B. Relative risks for any cancer other than breast or ovarian cancer in mutation carriers or populations eligible for mutation testing.
Eleven studies reported sufficient information to plot incidences of cancers lumped together in categories called “all cancers except breast, and ovarian” or “all cancers except breast, ovarian and nonmelanoma skin cancer. Figure 1B shows results for the reported incidences of cancers with statistically significant increases in relative risk of cancer at sites beyond breast or ovary. Results for different groups within each study were pooled together. Figure 1B shows that 5 of the studies do not include 1 in the confidence interval, and relative risks from all of the studies are > 1 with P values < .05. The combined risk was calculated as 1.52 (CI 1.21–1.93)
Most studies in Table 1 and Figures 1A and 1B demonstrated a statistically significant increased incidence for at least 1 cancer other than breast or ovarian cancer in BRCA gene carriers. This increased incidence crosses different methodologies and populations. This is consistent with the idea that the cancers occur sporadically but at moderately elevated frequency.
Figures 2A–D are forest plots of risks for stomach/gastric, pancreatic, prostate and colorectal cancer, respectively. These all show elevated relative risk. Surprisingly, the largest increase in relative risk was from stomach/gastric cancer (Figure 2A) RR = 1.69 [1.21–2.38] followed closely by pancreatic cancer (Figure 2C), RR = 1.62 [1.31–2.00].
Figure 2.
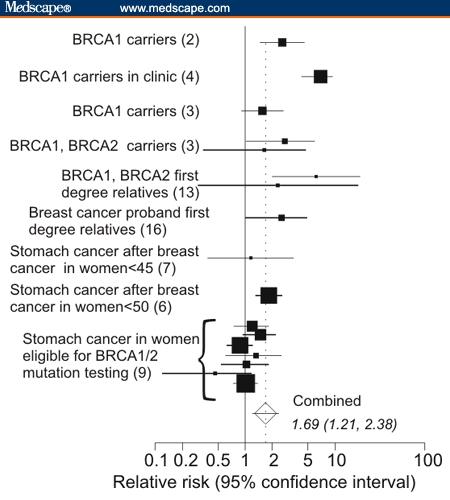
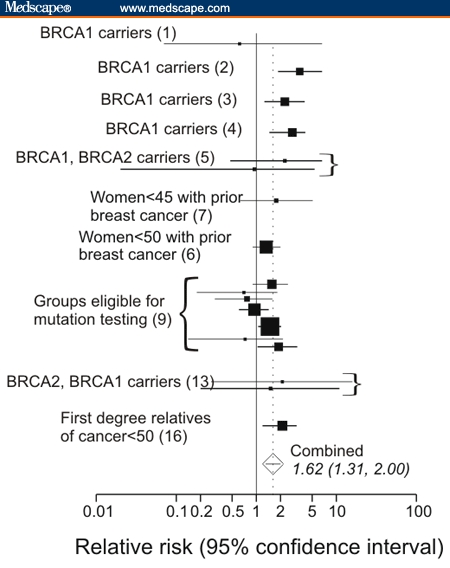
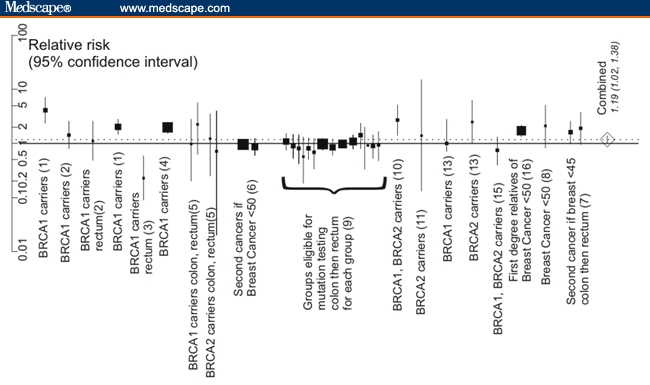
A. Relative risks of stomach/gastric cancer based on data in different population studies. B. Relative risks for prostatic cancer. C. Relative risks for pancreatic cancer. D. Relative risks for colon and for rectal cancer.
In Table 1, Studies 1-3 were from the Breast Cancer Linkage Consortium, a worldwide cooperative network of scientists and physicians with a major interest in inherited breast and ovarian cancer.[1–3] Study 2 contains a listing of cancers as “all cancers except breast, ovary, nonmelanoma skin and prostate cancer.” The odds ratio for these other, presumably sporadic cancers is 1.90.[2] Study 3 gives a relative risk of 1.34 [1.19–1.51] of all cancers except breast, ovary, nonmelanoma skin, prostate, and pancreatic cancer.[3]
Study 2 examined data from a large number of BRCA1 carriers.[2] Its increased statistical power detected moderate increases in numerous abdominal cancers (Table 1). In focusing on a clinic-based sample of BRCA mutation carriers,
Brose and colleagues[4] (Study 4, Table 1) found that colon, pancreatic, and gastric cancer occurred more frequently than in the general population. The cumulative age-adjusted risk of colorectal cancer in their study was a statistically significant 2-fold increase (P < .05). They also estimated a 3-fold increase in pancreatic cancer risk and a 4-fold increase in gastric cancer risk. The cumulative age-adjusted lifetime risk of any cancer other than breast and ovarian diagnosis in this group of BRCA1 mutation carriers was 13.8% (95% CI 10.7% to 16.9%). Of note, the overall risk of cancers other than of the female breast and ovary was statistically significantly higher for men than for women, with cumulative age-adjusted lifetime risks of 26.1% (95% CI 17.5% to 34.6%) and 10.3% (95% CI 7.2% to 13.3%), respectively.
A study of families related to BRCA1 probands from Sweden (Table 1, Study 5) reported nearly 6 times the risk of gastric cancer in females.[5] In a Japanese study (not listed in Table 1), a series of gastric cancer patients diagnosed before age 35 documented allelic losses flanking BRCA1 in 12 of 27 cases (44%), suggesting a possible mechanistic link between BRCA1 and the development of gastric cancer.[34]
Johansson and colleagues[5] (Table 1, Study 5) also found about a 6-fold increase in squamous cell skin cancer in males from BRCA1 families. Invasive cervical cancer was increased in females from BRCA2 families and prostate cancer was of borderline significance (Table 1, Study 5). Study 5 used expanded pedigrees based on mutation testing only for probands, so Study 5 would also include mutations elsewhere in BRCA pathways. Thus, effects such as amplification of EMSY genes are automatically included. EMSY overexpression may occur at least as often as BRCA mutations. A disadvantage of not typing mutations is that mutation carriers are still only a minority of the population and their risk values are diluted by including individuals with normal BRCA pathways. This makes it more difficult to show increased risks for other cancers.
The work by Evans and colleagues[6] (Table 1, Study 6) also did not directly type BRCA gene mutations. Evans and colleagues used data from the Thames Cancer Registry to identify women younger than 50 years of age who had additional primary cancers after breast cancer; 32,799 women were diagnosed with breast cancer younger than age 50 between January 1, 1961 and December 31, 1995. Women were censored from the study at clear cut-off dates. The mean follow-up period was 7.5 years. Within the 9 sites monitored, 1448 cases had multiple additional tumors; 1389 had 2 tumors, 57 had 3, and 2 had 4 tumors. On the basis of Myriad Genetics tables, the odds that a woman related to one of these individuals would have a BRCA mutation is only 7.3%. Also included in the study were 318 women who had multiple tumors when they were younger than 50. The figure of about 7.3% is far above that for the general population but still omits those with mutations elsewhere in BRCA pathways.
The early age of onset of breast cancer is considered characteristic of inherited cancers. Evans and colleagues[6] also provide data for women older than 50, and this permits useful comparisons. Comparing women with breast cancer diagnosed younger than age 50 with those diagnosed older than age 50, Evans and colleagues noted the younger age group had an increased risk of a second cancer at a site other than breast. Table 1 and Figures 1 and 2 show that the risk for a number of second cancers is elevated, and some of these rise to statistical significance.
Harvey and Brinton[7] (Table 1, Study 7), in a 1985 study, listed primary cancers after a first breast cancer in Connecticut women under age 45. The authors noted a significant relationship between breast and subsequent colon cancer. Second cancers were much more numerous in this group of nearly 7000 younger women than in older women. They found a significant downward trend for risk of colon cancer (RR = 1.6, 1.3, 1.1) and rectal cancer (RR = 1.9,1.1,1.0) with age. This downward trend applied to ages < 45, 45-54, and 55+. Similarly, Teppo and colleagues[8] (Table 1, Study 8) found that colon cancer in women after breast cancer was diagnosed before age 50 occurred almost twice as often as it did in women diagnosed with breast cancer after age 70.
Bermejo and Hemminki[9] used the Swedish Cancer Registry families to find families for which data existed for 3 generations. From these families, they selected individuals eligible for BRCA mutation testing. The results show an increased incidence of cancers in organs other than breast and ovary in a large population eligible for BRCA mutation testing. Cancers with increased standardized incidence ratios included prostate cancer, pancreatic cancer, and pancreatic cancer before age 50. Perhaps eye cancer and stomach cancer by age 70 also achieved statistical significance (Table 1, Study 9.)
Including this study was not without complications. There was no follow-up after the first additional cancer, so Bermejo and Hemminki would overlook slower-growing cancers such as colorectal cancers in favor of ovarian cancer and other faster-growing tumors. Ovarian and fallopian cancers were counted as an additional cancer, and the risk for these tumors varied from a few- to many-times greater than the risk for other cancers. In most groups, the odds were high that a diagnosis of ovarian cancer would have terminated follow-up, especially in the group defined by the presence of ovarian cancer in the family. Increased risk for stomach cancer did not rise to statistical significance until age 70 in 1 group. It is thus likely that including data of Bermejo and Hemminki leads to an underestimate of risk for other cancers, particularly slower-growing tumors such as colon cancer. Although nothing was excluded, excluding even some of the data of Bermejo and Hemminki (such as the group with ovarian and breast cancer in the family) would markedly increase the risk for colon cancer.
Study 10 used a different population and methodology but still supports the increased incidence of other cancers in BRCA mutation carriers. Shih and colleagues[10] (Study 10) found that 98 women with breast cancer who reported at least 1 other primary cancer in themselves or in a relative with breast cancer were twice as likely to be mutation carriers as women with breast cancer who did not report a second primary tumor. Their data support the notion that a general cancer susceptibility exists in those with BRCA1 and BRCA2 mutations, and a heightened clinical awareness is warranted in dealing with mutation carriers
Easton and colleagues[11] (Table 1, Study 11) studied 2 large breast cancer families with tumors linked to BRCA2 mutations. They found statistically significant increases in laryngeal cancer, prostate cancer, and ocular cancer. In other studies, there are implications that ocular cancer is elevated, but the disease is so rare that it is difficult to achieve statistical significance.
Moslehi and colleagues[12] (Table 1, Study 12) found relatives of Ashkenazi Jewish female BRCA2 carriers to be at greater lifetime risk (to age 75) for cancer of any type than the relatives of the BRCA1 carriers (46.3% vs. 34.9%). In men younger than 65 years, the risk was significantly higher for cancer (21.4% vs. 4.4 %;) and this was attributed largely to an excess of prostate, pancreatic, and colon cancers observed in male relatives of BRCA2 carriers at that age. In first-degree relatives of Jewish ovarian cancer patients, greater risks for pancreatic cancer, prostate cancer, and cancers in which the primary site was unknown were statistically significant.
In a population series of 649 women with ovarian cancer, Risch and colleagues[13] (Table 1, Study 13) examined BRCA2 mutation location relative to colorectal, stomach, pancreatic, and prostate cancer in family members. For probands carrying BRCA2 mutations, colorectal cancer in family members occurred only when mutations were within the ovarian cancer cluster region of exon 11 of the BRCA2 gene (now defined as nucleotides 4075 to 6503). Statistically significant increases in risk for colorectal, stomach, pancreatic, or prostate cancer (as well as for ovarian cancer) occurred for these mutations.
Aretini and colleagues[14] (Table 1, Study 14), as part of the Italian Consortium for Hereditary Breast and Ovarian Cancer, found that cancers other than breast or ovarian were significantly elevated in mutation carriers. The presence of prostate or pancreatic cancer in a family was correlated with the presence of ovarian cancer in BRCA2 mutation carriers.
Colon cancer is highly age dependent, rising from an incidence of 0.23% at age 50 to 5.6% at age 80. Because it was a peripheral issue, the publication did not list data to estimate the rate of colon cancer in a control group, so colon cancer incidence data had to be excluded. Nonetheless, the authors mentioned finding an increased incidence in BRCA families of pancreatic, prostate, and gastrointestinal cancers.
Streuwing and colleagues[15] (Table 1, Study 15) recruited over 5300 Jewish people in the Washington DC area and identified 120 carriers of 1 of the 3 BRCA founder mutations. The family histories of the carriers included increased percentages of numerous cancers other than breast or ovarian cancer. Although the numbers of carriers were low, there were elevations in prostate, lung, multiple myeloma, and Hodgkin's disease that reached sufficient statistical significance to warrant further investigation.
Goldgar and colleagues[16] (Table 1, Study 16) found a highly significant statistical association between familial occurrences of breast, colon, and prostate cancers and between breast and thyroid cancers. Berman and colleagues[17] in a small study (Table 1, Study 17) noted significant cancer histories in 8 BRCA2 mutation carriers.
The Occurrence of BRCA1 or BRCA2 Mutations in Subgroups of Patients With Other Cancers
Mutations are prevalent in familial forms of both pancreatic and prostate cancers when multiple relatives are affected. Carriers of BRCA1 or BRCA2 mutations represent up to 4% to 7% of all unselected pancreatic cancer patients and 10% of those from Ashkenazi Jewish families. In the Swedish cancer registry study of probable BRCA mutation carriers, the incidence of early onset pancreatic cancer was elevated up to 6.54-fold.[9]
With considerable variability, studies of patients presenting with pancreatic cancer or prostate cancer have concluded that BRCA1/BRCA2 mutation carriers are at increased risk for these cancers (Table 2, Studies 18–20). The studies in Table 2 generally support those in Table 1. Because of the low percentages of mutation carriers in the general population, only low numbers of mutation carriers were involved in the studies in Table 2, ranging from a high of 64 to a low of 3 (see column 4).
Pancreatic Cancer
Pancreatic cancer in BRCA2 mutation carriers is consistent with the possibility that BRCA2 loss sometimes promotes the malignant progression of existing lesions (Table 2). Familial pancreatic cancer marked by BRCA2 mutations occurs 8 to 10 years sooner than sporadic disease.[9,18,33] One explanation for the higher prevalence of pancreatic carcinoma in families with ≥ 2 affected first-degree BRCA2 mutation carriers is the early biallelic inactivation of the BRCA2 gene.[35] The risk for sporadic pancreatic cancer in BRCA2 mutation carriers rises to 3.5. Hahn and colleagues[18] studied 64 patients (37 men and 27 women) with pancreatic cancer among 26 families. Four families had 4 members with pancreatic cancer, 4 families had 3 affected members, and 18 families had 2 affected members; 3 families had 3 affected generations, 16 families had 2 affected generations, and 7 families had only 1 affected generation. No families were Ashkenazi Jews (Table 2, Study 18).
Murphy and colleagues[19] (Table 2, Study 19) found increased risk of both sporadic and familial pancreatic cancer in BRCA2 mutation carriers. Within these kindreds, both Hahn and colleagues[18] and Murphy and colleagues[19] found a variety of additional cancers within the families they studied, but risks were not calculated.
These 2 reports[18,19] suggest that some familial pancreatic cancers are caused by BRCA2 germline mutations. Of note, these pancreatic cancer families generally may not show an increased incidence of breast and ovarian cancer. Other pathways not mediated via BRCA proteins are also involved and apparently generate pancreatic cancers independent of BRCA proteins.[33]
Prostate Cancer
Kirchhoff and colleagues[21] (Table 2, Study 21) studied the incidence of 3 founder BRCA mutations in unselected prostate cancer patients. When results were stratified by gene, BRCA2 mutation carriers were at increased risk for developing prostate cancer (odds ratio 4.78; [1.87-12.25]. The risk of BRCA1 mutation carriers was not significantly increased. Vazina and colleagues (Study 23) found that the rate of founder BRCA1 mutations in Ashkenazi Jewish prostate cancer patients (2.3% and 3.3%) was greater than the rate of the general population (0.77% and 0.37%).[23] Mutation carriers had double the risk of the general Ashkenazi population. Only 1 of 5 carriers had prostate cancer detected early.
In the relatively homogenous Icelandic population, 1 of 2 Icelandic population studies found significant elevations in prostate cancer risk for BRCA2 mutation carriers (Table 2, Studies 25 and 26).[25,26]
Ocular Melanoma
Several studies of ocular melanoma patients also found significant elevations in BRCA1/2 mutation carriers (e.g., Study 27).[27] The incidence of ocular melanoma appears to be 2% to 3% in BRCA mutation carriers, but there is insufficient data published to include them in relative risk plots in Figures 1 and 2.
Colon Cancer
Studies by Neill and colleagues[29] and Kirchhoff and colleagues[30] do not exclude a small increase in risk for colon cancer. The 2 studies involved only a small number of mutation carriers (24 and 8 mutation carriers, respectively). Neill and colleagues found a modest elevation in colon cancer risk that did not rise to statistical significance: BRCA1 or BRCA2 mutation OR = 1.47 [0.76 to 2.83)], adjusted OR = 1.50 [0.77 to 2.95]. A graph of the risk of colon cancer vs. number of mutation carriers in all available studies (not shown) suggests that a much larger number of mutation carriers may be needed to demonstrate increased risk. Thiffault and colleagues[36] noted that it is not uncommon to find families with multiple cases of breast and colon cancer. They reported on 1 family with 10 cases of breast or colon cancer among 26 first-, second- or third-degree relatives. The family had an apparent dual mutation in both BRCA2 and MSH2.
Mechanistic Studies Suggesting BRCA Mutations are Involved in Other Cancers
Hughes-Davies and colleagues[37,38] reported that inhibition of BRCA function by EMSY amplification led to myelomas and liver cancer. Relationships among BRCA- and Fanconi anemia proteins[35] suggest involvement with leukemias under some conditions.
Discussion
The incidence of cancers other than breast or ovarian was a peripheral issue in most studies, but the high quality of the data makes the present analysis feasible. Although the sites that show increased risk for cancer may vary among studies, essentially all applicable studies support the idea that damage to BRCA-related pathways increases overall cancer risks beyond breast and ovarian (Table 1 and Figure 1). Figures 1 and 2 also show some specificity in the organs at risk because an increase in cancer susceptibility was found more frequently for the stomach, pancreas, prostate, and colon/rectum. The amount of the increase in risk ranges from about 20% to about 60%.
In comparison to breast and ovarian cancers, the frequencies of additional cancers associated with BRCA1 or BRCA2 pathway mutations are lower. These include some cancers that have a high incidence rate in the general population and are thus not unexpected in families with BRCA mutations. This makes it essential to review a large amount of data. Generally in comparing studies in Table 1 that included BRCA mutation testing, those studies which involved the greatest number of mutation carriers reported the largest number of different sites for cancers beyond breast and ovarian. The size of the effect can be debated in some tissues. But loss of BRCA-related pathways more generally favors tumor growth or improves tumor survival beyond the breast and ovary. Moreover, BRCA-related gene pathways function in numerous sites throughout the body, not just in breast or ovary.
Despite the interrelationships of BRCA1 and BRCA2,[35] different BRCA1/BRCA2 mutations could have different functional effects. Table 1 and Table 2 and Figures 1 and 2 suggest that either mutation increases risk for a variety of cancers. Many studies tested only for BRCA1 or BRCA2 founder mutations and were unable to detect mutations of different types, which now make up a substantial minority of the total set. None of the studies tested for EMSY overexpression, because EMSY was unknown at the time of the studies. In significant percentages of sporadic breast and ovarian cancers, overexpression of EMSY may be functionally equivalent to loss of BRCA genes. Heterozygous ATM mutations have also been associated with increased risk for breast cancer and potentially for other cancers as well.[39] Thus, the risk for a variety of cancers can be evaluated by considering the whole BRCA pathway, not just BRCA1/2 genes. These arguments also justify this report's use of data not only from BRCA mutation carriers but also from people eligible for BRCA mutation testing. The percentages of the population with a defect in the BRCA pathway other than in BRCA genes may be significantly larger than the percentages of people with BRCA1 and BRCA2 gene mutations. This implies that other components of BRCA pathways may be targeted in some sporadic cancers. This properly shifts the focus from an individual gene to the pathway or the local area of the network in which the gene resides.
Other inherited gene mutations, including those in PTEN and p53, also predispose to breast cancer and would contribute to relative risks for other cancers. These mutations have a rarer incidence than those in BRCA genes, and a case can be made that their pathways have some relationship to those involving BRCA1 and BRCA2.
Sporadic cancers are consistent with known mechanisms of BRCA gene actions.
A generalized susceptibility to a variety of additional tumors is consistent with current ideas of BRCA1 and BRCA2 functions. Figure 3 shows the evolving diagram of proposed functions and interactions among BRCA-mediated gene pathways. The figure summarizes published functional implications and models of DNA damage responses involving BRCA1 and BRCA2. Mechanisms that sense DNA damage activate protein kinases such as ATM or ATR, triggering a cascade of phosphorylation-dependent steps leading to cell-cycle checkpoint arrest, DNA repair, or apoptosis. BRCA1, BRCA2, and Fanconi anemia (FA) proteins are involved in these steps. The MRE-Rad50-Nbs1 complex indicated in the Figure also functions as a double strand break sensor that recruits ATM to broken DNA molecules.[40] Figure 3 stresses that BRCA1 and BRCA2 are involved in pathways that regulate DNA repair, cell-cycle progression, ubiquitylation, and apoptosis.
Figure 3.
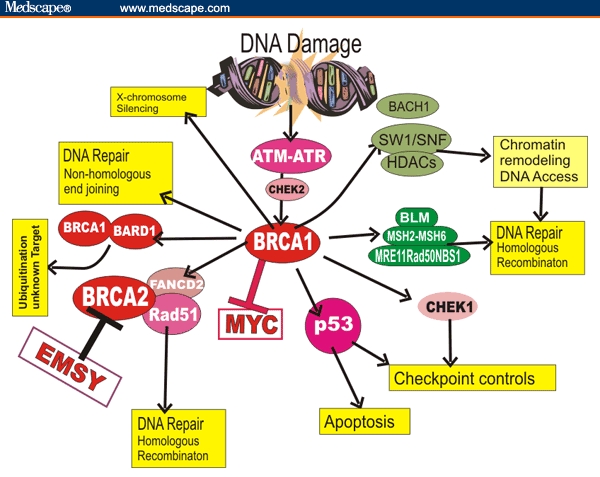
Examples of interactions involving BRCA1 and BRCA2 proteins stressing the general nature of their functions and some interactions with BRCA1. The exact order of events and understanding of these interactions are still evolving.
Inhibition of MYC action by interacting with BRCA1 may help explain why loss of BRCA function impacts some sporadic cancers. BRCA1-IRIS or unrecognized factors may participate in determining tissue specificity to limit BRCA influences in other tumors or they may significantly alter our current understanding. Moreover some relationships and interactions in Figure 3 are likely to occur only in specific physiologic contexts. For example, MYC overexpression or BRCA mutation may have different effects depending on when in development they occur because the genomic background is different.[38] Nonetheless, the above considerations may be helpful in understanding involvement of BRCA-related pathways in limiting tumors in organs other than breast or ovary. The above pathways and functional associations help explain the documented clinical associations.
It is possible that different populations in different geographical areas may have different susceptibilities. Sporadic cancers are sensitive to diet and environmental exposure, and these cannot be controlled in any observational study. The possibility that chemotherapy for breast or ovarian cancers induces secondary tumors more readily in mutation carriers cannot be excluded. Standard chemotherapy and radiation regimens for breast cancer would increase risks for leukemia and other cancers.[10] Risk for (myeloid) leukemia is statistically significantly elevated in the Evans study[6] but not in the Bermejo study.[9] Goldgar and colleagues[16] and Harvey and Brinton[7] did not find an increased risk of leukemia associated with breast cancer.
One might expect that BRCA mutations would lower the age of onset of sporadic cancers. There are limited data on the age at onset of sporadic cancer vs. incidence of other cancers. Harvey and Brinton[7] found a significant downward trend for risk vs. age for colon cancer (RR = 1.6, 1.3, 1.1) and rectal cancer (RR = 1.9,1.1,1.0) for ages < 45, 45–54, and 55+. This is consistent with BRCA mutations affecting the incidence/onset of colon cancer.
A lowered age at onset may be difficult to demonstrate more convincingly, particularly for slow-growing cancers such as colon cancer. It is likely that screening practices vary widely among population groups within individual studies. Patients usually have no symptoms or only nonspecific ones until their cancers reach a large size or spread to other organs. Current colon cancer screening misses many early cases,[41] up to almost 85% by fecal occult blood testing. Sigmoidoscopy may miss over 92% of advanced colorectal neoplasia in the proximal colon.[42] Screening for colon cancer is notorious for its poor compliance. BRCA mutations occur with comparatively high frequency in Israel; yet in 2000, almost half the Israeli population was unaware of colorectal cancer screening, and only 38% had any interest in participating. Thus, many cases of the disease are detected only at an advanced stage.[41] Colon cancer takes 20 to 40 years to develop with no intermediate endpoints to measure rate of development after transformation. Long follow-up was not frequently done in the studies reviewed so that slow-growing tumors might be missed.
Some cancers (such as prostate cancer) in BRCA mutation carriers or those eligible for mutation testing may not have distinct histopathology. Despite the BRCA-relationship, some routes of cancer evolution could still be abolished because some checkpoint mutations are lethal and limit diversity. The initiating oncogenic events may restrict ensuing pathways to limit the subsequent mutation profile of the cancers that occur, pruning disease entities so that the surviving tumors do not differ histologically from sporadic cancers.
References
- 1.Ford D, Easton D, Bishop T, Narod S, et al. Risks of cancer in BRCA1 mutation carriers. Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 2.The Breast Cancer Linkage Consortium. Cancer risks in BRCA1 mutation carriers. J Natl Cancer Inst. 1999;91:1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 3.Thompson D, Easton DF. Breast Cancer Linkage Consortium. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 4.Brose M, Rebbeck T, Calzone K, et al. Cancer risk estimates for BRCA1 mutation carriers identified in a cancer risk evaluation program. J Natl Cancer Inst. 2002;94:1359–1365. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- 5.Johannsson O, Loman N, Moller T, Kristoffersson U, Borg A, Olsson H. Incidence of malignant tumors in relatives of BRCA1 and BRCA2 germ-line mutation carriers. Eur J Cancer. 1999;35:1248–1257. doi: 10.1016/s0959-8049(99)00135-5. [DOI] [PubMed] [Google Scholar]
- 6.Evans H, Lewis C, Robinson D, Bell C, Moller H, Hodgson S. Incidence of multiple primary cancers in a cohort of women diagnosed with breast cancer in southeast England. Br J Cancer. 2001;84:435–440. doi: 10.1054/bjoc.2000.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harvey EB, Brinton LA. Second cancer following cancer of the breast in Connecticut, 1935–1982. Natl Cancer Inst Monogr. 1985;68:99–109. [PubMed] [Google Scholar]
- 8.Teppo L, Pukkala E, Saxen E. Multiple cancer – an epidemiological exercise in Finland. J Natl Cancer Inst. 1985;75:207–217. [PubMed] [Google Scholar]
- 9.Bermejo JL, Hemminki K. Risk of cancer at sites other than the breast in Swedish families eligible for BRCA1 or BRCA2 mutation testing. Ann Oncol. 2004;15:1834–1841. doi: 10.1093/annonc/mdh474. [DOI] [PubMed] [Google Scholar]
- 10.Shih H, Nathanson K, Seal S, Collins N, Stratton M, Rebbeck T, Weber B. BRCA1 and BRCA2 mutations in breast cancer families with multiple primary cancers. Clin Cancer Res. 2000;6:4259–64. [PubMed] [Google Scholar]
- 11.Easton D, Steele L, Fields P, Ormiston W, et al. Cancer risks in two large breast cancer families linked to BRCA2 on Chromosome 13q12-13. Am J Hum Genet. 1997;61:120–128. doi: 10.1086/513891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moslehi R, Chu W, Karlan B, et al. BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet. 2000;66:1259–1272. doi: 10.1086/302853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Risch H, McLaughlin J, Cole D, Rosen B, et al. Prevalence of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68:700–710. doi: 10.1086/318787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aretini P, D'Andrea E, Pasini B, Viel A, et al. Different expressivity of BRCA1 and BRCA2: analysis of 179 Italian pedigress with identified mutation. Breast Cancer Res Treat. 2003;81:71–79. doi: 10.1023/a:1025428807472. [DOI] [PubMed] [Google Scholar]
- 15.Streuwing J, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 16.Goldgar DE, Easton DF, Cannon-Albright L, Skolnick M. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86:1600–1608. doi: 10.1093/jnci/86.21.1600. [DOI] [PubMed] [Google Scholar]
- 17.Berman D, Costalas J, Schultz D, Grana G, Daly M, Godwin A. A common mutation in BRCA2 that predisposes to a variety of cancers is found in both Jewish and non-Jewish individuals. Cancer Res. 1996;56:3409–3414. [PubMed] [Google Scholar]
- 18.Hahn S, Greenhalf B, Ellis I, Sina-Frey M, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95:214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- 19.Murphy K, Brune K, Griffin C, Sollenberger J, et al. Evaluation of candidate genes MPP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17% Cancer Res. 2002;62:3789–3793. [PubMed] [Google Scholar]
- 20.Lal G, Liu G, Schmocker B, et al. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1 and BRCA2 mutations. Cancer Res. 2000;60:409–416. [PubMed] [Google Scholar]
- 21.Kirchhoff T, Kauff N, Mitra N, et al. BRCA mutations and the risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res. 2004;10:2918–2921. doi: 10.1158/1078-0432.ccr-03-0604. [DOI] [PubMed] [Google Scholar]
- 22.Edwards SM, Kote-Jarai Z, Meitz J, et al. Two percent of men with early onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet. 2003;72:1–12. doi: 10.1086/345310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazina A, Baniel J, Shtriker A, et al. The rate of the founder Jewish mutations in BRCA1 and BRCA2 in prostate cancer patients in Israel. Br J Cancer. 2000;83:463–466. doi: 10.1054/bjoc.2000.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubert A, Peretz T, Manor O, et al. The Jewish Ashkenazi founder mutations in the BRCA1/BRCA2 genes are not found at an increased frequency in Ashkenazi patients with prostate cancer. Am J Hum Genet. 1999;65:9212–924. doi: 10.1086/302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorlacius S, Struewing J, Hartge P, et al. Population based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet. 1998;352:1337–1339. doi: 10.1016/s0140-6736(98)03300-5. [DOI] [PubMed] [Google Scholar]
- 26.Sigurdsson S, Thorlaciaus S, Tomasson J, et al. BRCA2 mutation in Icelandic prostate cancer patients. J Mol Med. 1997;75:758–761. doi: 10.1007/s001090050162. [DOI] [PubMed] [Google Scholar]
- 27.Iscovich J, Abdulrazik M, Cour C, et al. Prevalence of the BRCA2 6174 del T mutation in Israeli uveal melanoma patients. Int J Cancer. 2002;98:42–44. doi: 10.1002/ijc.10155. [DOI] [PubMed] [Google Scholar]
- 28.Scott R, Vajdic C, Armstrong B, et al. BRCA2 mutations in a population based series of patients with ocular melanoma. Int. J Cancer. 2002;102:188–191. doi: 10.1002/ijc.10693. [DOI] [PubMed] [Google Scholar]
- 29.Niell B, Rennert G, Bonner J, Almog R, Tomsho L, Gruber S. BRCA1 and BRCA2 founder mutations and the risk of colorectal cancer. J Natl Cancer Inst. 2004;96:15–21. doi: 10.1093/jnci/djh008. [DOI] [PubMed] [Google Scholar]
- 30.Kirchhoff T, Satagopan J, Kauff N, et al. Frequency of BRCA1 and BRCA2 mutations in unselected Ashkenazi Jewish patients with colorectal cancer. J Natl Cancer Inst. 2004;96:68–70. doi: 10.1093/jnci/djh006. [DOI] [PubMed] [Google Scholar]
- 31.Chen-Stoyerman R, Figer A, Fidder H, et al. The frequency of the predominant Jewish mutations in BRCA1 and BRCA2 in unselected Ashkenazi colorectal cancer patients. Br J Cancer. 2001;84:475–477. doi: 10.1054/bjoc.2000.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drucker L, Stackievitz R, Shpitz B, Yarkoni S. Incidence of BRCA1 and BRCA2 mutations in Ashkenazi colorectal cancer patients: preliminary study. Anticancer Res. 2000;20:559–561. [PubMed] [Google Scholar]
- 33.Bardeesy N, DePinho R. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 34.Semba S, Yokozaki H, Yasui W, Tahara E. Frequent microsatellite instability and loss of heterozygosity in the region including BRCA1 (17q21) in young patients with gastric cancer. Int J Oncol. 1998;12:1245–1251. doi: 10.3892/ijo.12.6.1245. [DOI] [PubMed] [Google Scholar]
- 35.Howlett N, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 36.Thiffault I, Hamel N, Pal T, et al. Germline truncating mutations in both MSH2 and BRCA2 in a single kindred. Br J Cancer. 2004;901:483–491. doi: 10.1038/sj.bjc.6601424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115:523–535. doi: 10.1016/s0092-8674(03)00930-9. [DOI] [PubMed] [Google Scholar]
- 38.Haber D. The BRCA2-EMSY connection: Implications for breast and ovarian tumorigenesis. Cell. 2003;115:507–512. doi: 10.1016/s0092-8674(03)00933-4. [DOI] [PubMed] [Google Scholar]
- 39.Thompson D, Duedal S, Kirner J, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 40.Lee J-H, Paull T. Double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 41.Mariani S. Conference Report – Early Cancer Diagnosis: Beating the Odds (CME); Highlights of the Annual Meeting of the 95th American Association for Cancer Research; March 27-31, 2004; Orlando, Florida. MedGenMed Hematology-Oncology. Medscape General Medicine. 2004;6(3) [PMC free article] [PubMed] [Google Scholar]
- 42.Schoenfeld P, Cash B, Flood A, et al. Colonoscopic screening of average-risk women for colorectal neoplasia. N Engl J Med. 2005;352:2061–2068. doi: 10.1056/NEJMoa042990. [DOI] [PubMed] [Google Scholar]