

Abstract
Control of nonspecific protein adsorption is very important for the design of biocompatible and biomimetic materials as well as drug carriers. Grafted polymer layers can be used to prevent protein adsorption. We have studied the molecular factors that determine the equilibrium and kinetic control of protein adsorption by grafted polymer layers. We find that polymers that are not attracted to the surface are very effective for kinetic control but not very good for equilibrium reduction of protein adsorption. Polymers with attractions to the surface show exactly the opposite behavior. The implications for molecular design of biocompatible materials also are discussed in this paper.
Protein adsorption plays a major role in a variety of important biological-related processes. Biocompatible materials are required to eliminate, or largely reduce, the adsorption of blood proteins, for example, to avoid surface-induced thrombosis (1, 2). In recent years special attention has been given to the modification of protein–surface interactions using grafted polymers (3, 4, 5, 6). The idea, borrowed from many years of research in colloidal stabilization (7) and mimicking one of the roles of polysaccharides in cell membranes (8), is to build a steric barrier to the proteins by the presence of the polymer layer (9, 10). In practice, however, there is still no systematic way of modifying surfaces that are successful in preventing protein adsorption. This is because of the complexity involved in the process caused by the interplay between changing average structure of the polymer–protein layer, strong protein–surface attractions, and polymer–surface interactions (11). For example, liposomes† formed by mixtures of lipid and lipid-polymer have shown increased longevity in the bloodstream and are now being used as drug delivery systems (12, 13). It is believed that the role of the polymers tethered to the liposome interface is to present a steric barrier to approaching proteins from the blood. However, the amount of polymer on the surface is rather small, and experimental observations of the same polymer grafted on hydrophobic surfaces, at the same surface coverage, show that although the amount of protein adsorbed is lower than in the absence of polymer, there is still a significant amount of protein adsorption (14).
In this paper we demonstrate that the molecular factors that determine the ability of the polymer layer to prevent protein adsorption are different for the kinetic control as compared with the equilibrium case. We will demonstrate that our theoretical predictions can quantitatively reproduce experimental observations of protein adsorption isotherms. We will show that surface–polymer interactions play a central role in the ability of the polymer layer to prevent protein adsorption. However, the role of those interactions is different for kinetic as compared with equilibrium control. Our findings enable us to rationalize why the surface-modified liposomes have an increased longevity but polymers on hydrophobic surfaces are not as effective to prevent protein adsorption. Finally, our theoretical predictions can be used to decide the type of surface modification necessary for the rational design of biomaterials.
Consider a protein solution of density ρb that is put into contact with a surface. The surface has polymers that are chemically attached to it at one of their ends. The presence of the surface induces a gradient in the chemical potential of the protein. This gradient arises from the sudden inhomogeneous environment that appears in the direction perpendicular to the surface. The proteins in the close vicinity of the surface feel the bare attractive interaction of the surface as well as the repulsions induced by the polymer molecules. The balance between these interactions will result in a final equilibrium density profile and amount of protein adsorbed.
The time evolution from the homogeneous system to the new equilibrium induced by the presence of the modified surface can be described (in the free-draining limit) with the help of a diffusion equation (15–18) of the form
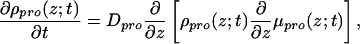 |
1 |
where we have assumed that the diffusion in the xy plane is faster than the adsorption. This assumption is justified for the relatively low surface densities that we treat here (19). The time-dependent local chemical potential is defined by μpro(z;t) =  , where W is the free-energy density of the system. This free energy includes the contribution of the grafted polymer molecules, the solvent, and the surface, under the condition of the fixed protein concentration profile given by ρpro(z;t) at each time t.
, where W is the free-energy density of the system. This free energy includes the contribution of the grafted polymer molecules, the solvent, and the surface, under the condition of the fixed protein concentration profile given by ρpro(z;t) at each time t.
The adsorption kinetics can be described with the help of Eq. 1, assuming that the time scale of the diffusion of the proteins is much slower than that of the polymer and solvent rearrangement. This is a physically motivated approximation, because the solvent molecules (water) are much faster than the protein motion, and the rearrangements of the polymer molecules involve only local moves, which, for the flexible chains of interest here, are much faster than the typical motion of the larger protein molecules.
The free energy of the system is determined by using a molecular mean-field theory. The predictions of the theory for equilibrium systems have been shown to be in excellent agreement with experimental observations for conformational and thermodynamic properties of tethered polymer layers (20, 21) as well as for the adsorption of proteins on hydrophobic surfaces (11, 14). The basic idea of the theory is to treat each molecule with all of its intramolecular and surface interactions exactly taken into account (within the chosen model system). The intermolecular interactions are considered within a mean-field approximation. The mean field is determined by the average properties of all of the molecules in the mixture, and thus the theory is in essence a self-consistent approach.
We write the free energy of the system in complete analogy with the equilibrium approach (22) but with all of the quantities now assumed to be time-dependent. The explicit expression for the time-dependent free energy density (per unit area) reads
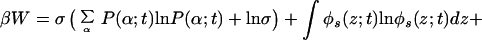 |
2 |
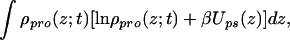 |
where the first two terms are the contribution from the tethered polymers, with the first being the conformational entropy of the chains, with P(α;t) being the time-dependent probability distribution function (pdf) of chain conformations and the second being the tethered polymers two-dimensional translational entropy. The third term is the solvent entropy, with φs(z;t) being the volume fraction of solvent at distance z from the surface at time t. The last two terms correspond to the translational entropy of the proteins and the protein–surface interaction, respectively, Ups(z) is the bare surface–protein interaction. This interaction is taken from ref. 23 where the potential between lysozyme and a hydrophobic surface was calculated by using atomistic potentials. Note that the solvent and protein contributions are integrated over z to account for all of the molecules.
Eq. 2 assumes that the intermolecular attractions between all of the components in the system are the same. This is actually the model that we successfully used to compare experimental observations and the predictions of the theory in early work (14) and in the comparisons shown below. The excellent agreement between the predictions of the theory and the experimental observations supports the assumption.
Eq. 2 does not include the repulsive interactions between the molecules. The hard-core repulsions are accounted for by packing constraints in which the available volume to the molecules, at each distance z from the surface, is filled by polymer, protein, or solvent molecules. This constraint reads
 |
3 |
where the first term is the volume fraction of polymer at z, with 〈ng(z;t)〉dz = 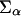 P(α;t)ng(z,α;t)dz being the average number of polymer segments of volume v0 at time t at distance z from the surface. The second term is the volume fraction of proteins with v(z;z′)dz being the volume that the proteins at distance z′ from the surface contribute to z. The last term is the volume fraction of solvent.
P(α;t)ng(z,α;t)dz being the average number of polymer segments of volume v0 at time t at distance z from the surface. The second term is the volume fraction of proteins with v(z;z′)dz being the volume that the proteins at distance z′ from the surface contribute to z. The last term is the volume fraction of solvent.
The assumption of solvent and polymer equilibration in the time scale of protein motion implies that the time-dependent pdf of tethered polymer configurations and the solvent-density profile are those that minimize the free energy (Eq. 2) subject to the packing constraint (Eq. 3) for fixed protein density profile. Thus, at each time t the density of proteins calculated from the diffusion equation (Eq. 1) is given as input to the free energy and this free energy is minimized subject to the packing constraints. This minimization is done by introducing time-dependent Lagrange multipliers, βπ(z;t). The pdf of chain conformations is
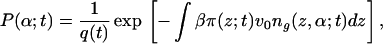 |
4 |
where q(t) is the time-dependent normalization constant. For the solvent-density profile, the expression is
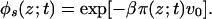 |
5 |
The numerical values of the Lagrange multipliers are obtained by replacing the expression for the pdf, Eq. 4, and the solvent-density profile, Eq. 5, into the constraint equation, Eq. 3, where the protein contribution is given as an input obtained from the diffusion equation, Eq. 1. Once the Lagrange multipliers are obtained, one can calculate the time-dependent protein chemical potential that is needed for the time evolution of the adsorption. This calculation is done by differentiating the free-energy density to obtain
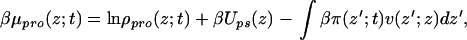 |
6 |
which is readily obtained once the Lagrange multipliers are known. Thus, the time evolution of the system is obtained in the following way. At time t = 0, a tethered polymer layer in equilibrium with pure solvent is brought into contact with a protein solution. The initial density profile of proteins enables the calculation of π(z;0) from which μpro(z;0) is obtained by use of Eq. 6. From it, the diffusion equation (Eq. 1) is iterated a time step. The new density profile of proteins now is given as input for the calculation of the new Lagrange multipliers from which the chemical potential profile is obtained and the time evolution is further iterated. This procedure is continued until the new equilibrium condition is achieved in which μpro(z;t) = μpro.eq for all z.
Eq. 6 shows that the motion of the proteins toward the surface contains three contributions. The first one is the purely diffusive motion that is found in any ideal system because of the gradient in composition. The second arises from the force exerted to the proteins by the surface. These two terms do not depend on the intermolecular interactions and will lead to a fast approach of the proteins to the surface. We refer to them as the purely diffusive motion (see e.g., Fig. 2 and discussion thereafter). The last term is the one arising from the intermolecular (protein–polymer and protein–protein) interactions. This term will be responsible for steric barriers and it is expected to lead to regimes in which the motion is dominated by kinetic barriers. The interplay between the diffusion and barrier crossing to determine the total adsorption process is discussed in detail below.
Figure 2.

(A) Amount of proteins adsorbed, in molecules per Å2, as a function of time. The dashed lines correspond to the equilibrium amount of protein adsorbed. The lines correspond to the following number of EO units: black, no polymer; red, n =6; green, n =15; blue, n = 25; and orange and magenta, n = 50. The magenta curve corresponds to a hydrophobic surface. (B) The potential of mean force, in kJ/mol, as a function of the distance from the surface for: black, bare protein–surface interaction; and red, n = 50 before the beginning of the adsorption. The green and blue curves are for n = 50 at the times marked by the arrows in A. (C) Maximal steric repulsion for n = 50 as a function of time. Note the changes in the shape and strength of the potential as the adsorption process takes place. All of the results correspond to a grafted density of σ = 0.0012Å−2. Higher surface coverage of polymer will result in slower kinetics but qualitatively similar behavior.
Fig. 1 shows the predicted equilibrium adsorption isotherms for lysozyme and fibrinogen on surfaces with short oligomeric polyethylene oxide (PEO) together with the experimental observations of Prime and Whitesides (24, 25). The calculations were carried out by full minimization of the free energy (thermodynamic equilibrium), i.e., with respect to the protein degrees of freedom as well, as explained in detail in refs 14 and 22. The agreement between the theory and the experimental observations is excellent. The adsorption isotherms show that as the surface coverage of polymer increases, the amount of protein adsorbed decreases. There is a surface coverage of oligomers above which there is zero protein adsorption. The role of the short chains is basically to fill the space close to the surface, reducing the amount of available surface for the adsorbing proteins (11, 22, 24, 25). The most important point to note is that these chains are short and that the surface coverage at which the adsorption goes to zero is very large. Those surface coverages are an order of magnitude larger than are typical surface coverages obtained by grafting PEO-2,000 or PEO-5,000‡. Further, the calculations assume that the ethylene oxide (EO) oligomers are not attracted to the surface. In contrast, the ability of long-grafted PEO to reduce protein adsorption on modified hydrophobic glass surfaces was found to be closely linked to the fact that the EO segments are strongly attracted to the surface and compete with the proteins for adsorption sites (14).
Figure 1.

Theoretical predictions for the amount of protein adsorbed as a function of the surface coverage of EO oligomers (full lines). The experimental observations (symbols) are from Prime and Whitesides (24, 25), where they measured the thickness of the adsorbed film (in nm), which is proportional to the surface density of protein, as a function of composition of a mixed self-assembled film. The mixed films are composed by SC10OH and SC11(EO)n, with n = 6 and 17, on gold surfaces. The relationship between the experimentally observed thickness and the protein density was fitted so that at zero composition the predictions and the experimental observations agree. The parameters used in the calculations are from refs. 11 and 22.
How good is the equilibrium assumption? To this end, we solve the diffusion Eq. 1 with the free energy obtained from the same theoretical approach that was used in the equilibrium predictions of Fig. 1. Fig. 2A shows the time evolution of the amount of protein adsorbed on the surface for a variety of PEO chain lengths. All of the profiles but one correspond to PEOs that are not attracted to the surface; we discuss the case of the attractive surface later. There are several interesting features that can be observed from the profiles. First, as the chain length of the grafted polymers increases up to 25 segments (corresponding close to PEO-1,000), the equilibrium amount of protein adsorbed decreases. However, for PEO-2,000, the amount of protein adsorbed at equilibrium is almost identical to that of PEO-1,000. The equilibrium amount of protein adsorbed is independent of polymer chain length once the thickness of the layer is larger than the protein size (22).
Second, the kinetics of protein adsorption is qualitatively similar for all chain lengths up to PEO-1,000. There are two qualitatively different regimes. For short times, the adsorption is very fast, and it is controlled by the diffusion of the proteins to the surface, i.e., the first two terms of Eq. 6. The second regime is a much slower one, and it is controlled by the repulsive barrier that the proteins already adsorbed on the surface and the grafted polymers present to the approaching proteins, i.e., the motion is dominated by the last term in Eq. 6. The total time to reach equilibrium in these cases supports the equilibrium assumption used to obtain Fig. 1.
Third, for PEO-2,000, there is no fast regime, i.e., there is no diffusion-controlled adsorption. At this molecular weight and for the surface coverage shown, the polymer chains present a steric barrier that causes the adsorption to be controlled by the kinetics of barrier crossing, Fig. 2B.
The dependence of the time scales for adsorption on polymer molecular weight also can explain the success of pegolated liposomes having increased longevity in the bloodstream. Typical molecular weights used in those systems are PEO-2,000 to PEO-5,000. As we can see from Fig. 2 for PEO-2,000, the chains are long enough that the adsorption is completely controlled by the kinetic barriers presented by the polymers, and the time scale to achieve equilibrium is much longer. Actually, the system only reaches equilibrium in many hours. Moreover, the calculations presented here are for lysozyme, which is a relatively small protein as compared to fibrinogen. Thus, for fibrinogen the time scale for adsorption is going to be much longer, because its large size results in very large repulsions caused by the interactions with the polymers before reaching the surface§.
The time-dependent adsorption for the case of PEO-2,000 with attractive interactions with the surface (hydrophobic surfaces) shows two important differences with the nonattractive case. First, the equilibrium amount of adsorbed proteins is much lower for the hydrophobic surface as compared with the same molecular weight for nonattractive surfaces. Namely, grafted polymers that are attracted to the surface are more effective in reducing protein adsorption. Second, the time scale for adsorption, both for the initial adsorption and to reach equilibrium, are orders of magnitude faster for surfaces with attractive interactions with the polymers. The reason for the faster kinetics is that the polymers have almost all of their segments close to the surface and thus present no steric barrier to the approaching proteins up to the point of close vicinity with the surface. The reason for being better at preventing the equilibrium adsorption is that the proteins need to compete for adsorption sites with the polymers. The short time scale for adsorption on hydrophobic surfaces suggests that the recent comparisons (14) with experimental observations assuming equilibrium are probably very good.
We now discuss in some detail the origin and characteristics of the repulsive barriers that the proteins feel in the different cases. First, let us consider the case of no grafted polymer or polymer molecular weights below PEO-2,000. For the surface coverage considered here, we see that in all of the cases there is a diffusion-controlled regime of the adsorption kinetics. This finding implies that the proteins approaching the surface feel a purely attractive interaction potential with no barriers. The dramatic change in slope in the kinetics is attributable to the presence of a barrier in the potential of mean force that the proteins approaching the surface feel¶. For the hydrophobic surfaces, the same effect is observed but up to much longer molecular weights (see above). For the PEO-2,000 with no attraction with the surface, the polymers present a kinetic barrier even in the absence of any adsorbed proteins (Fig. 2 B and C). The presence of the barrier implies that there is no diffusion-controlled regime. Furthermore, any molecular weight above 2,000 will show a stronger barrier and thus the time scale of the initial adsorption will be even longer.
The main conclusion from this work is that the best surface modifiers for the kinetic control of protein adsorption are polymers that are not attracted to the adsorbing surface. However, the best type of polymers for the thermodynamic control of protein adsorption are those that are attracted to the surface. The molecular picture that emerges is summarized in Fig. 3, where a cartoon is shown of how the different types of polymers kinetically or thermodynamically prevent the protein adsorption, along with the calculated variation of the shape of the grafted polymers as a function of time. The complexity involved in the adsorption process can be seen in the time-dependent average shape of the polymer molecules. The kinetics of protein adsorption on surfaces with grafted polymers involves the competition between the strong bare attractive interactions between the surface and the proteins and the conformational entropy loss that the grafted polymers have to pay to accommodate the proteins. This process involves a constantly varying environment, and therefore the kinetic behavior cannot be characterized in simple terms. We find that the kinetic barriers for protein adsorption vary constantly in time, and the whole potential is a complicated function that varies with distance and time as a result of the constant rearrangement of the polymer–protein layer (see Fig. 2 B and C).
Figure 3.

(Left) Schematic representation of the ability of grafted polymers to prevent protein adsorption. For polymers attracted to the surface, there is no kinetic barrier but the protein competes with the polymer for adsorption sites. Polymers that are not attracted to the surface present a large steric barrier but not very good thermodynamic prevention because of the ability of the protein to deform the polymer layer. (Right) The curves on the right show the calculated average shapes of two neighboring polymers as a function of time. The shape is defined here (20) by the lateral average radius of gyration of the chains as a function of the distance from the surface. Also, a protein is shown to scale, demonstrating that the deformation of the polymer layer is exactly what is needed for the protein to reach the surface. The black curves correspond to t = 0, the green and blue curves are for the times marked by the same color arrows in Fig. 2A, and the red curve is the final equilibrium state.
We have presented a detailed study of the equilibrium and kinetic adsorption of proteins on surfaces with grafted polymers. The extension of the molecular theory for tethered polymer layers to dynamical systems enables the study of kinetic processes over several orders of magnitude in time without compromising molecular detail. Note that although this approach does not provide the detail of molecular dynamics simulations, it enables the study of the dynamic processes over 15 orders of magnitude in time, thus bridging the gap between microseconds and days in complex systems with the ability to follow changes on average molecular properties.
Biocompatible materials can be in contact with the bloodstream for relatively short times (e.g., drug carriers) or very long times (e.g., artificial organs). The type of surface modifier that is necessary for protein rejection in each of these cases is different. In systems where it is necessary to control protein adsorption over finite time scales, it is probably best to use relatively dense polymer layers with long polymers that are not attracted to the surface of the biomaterial. This scenario should provide the best kinetic control. On the other hand, for materials that need to be in contact with the bloodstream for years, the optimal type of polymers may be one that is attracted to the surfaces, then providing thermodynamic control. It is important to mention that very high surface coverage of grafted polymers is extremely hard to obtain experimentally (14, 26). Thus, alternative approaches may be needed, for example, mixtures of polymers that present optimal kinetic and thermodynamic control under experimental realizable conditions. The theory used here provides quantitative guidelines on how to build those optimal layers.
Acknowledgments
This work is supported by the National Science Foundation through Grant CTS-9624268. I.S. is a Camille Dreyfus Teacher/Scholar.
Abbreviations
probability distribution function
- EO
ethylene oxide
- PEO
polyethylene oxide
Footnotes
Liposomes are closed spherical bilayers formed by lipid molecules with varying radii from 10 nm to micrometers.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.150236197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.150236197
PEO-M, also called PEG-M for polyethylene glycol, is a chain of molecular weight M. Thus, the number of segments in the chain is given by M/45. We refer here to the commonly used experimental molecular weights. The exact number of segments used in the calculations is given in the figure legends.
The bare potential of fibrinogen with surfaces is not known and therefore it is very hard to provide a reliable estimate for the potential of mean force. The calculations presented here use the same attractive contribution between the surface and fibrinogen that was proposed in ref. 14. This interaction is only the attractive strength at contact.
The potential of mean force here refers to the interaction (as a function of the distance from the surface) of a protein with the surface averaged over all of the configurations of the polymer molecules, solvent, and other proteins in the system, which is given exactly by the last term in Eq. 6.
References
- 1.Horbett T A. Cardiovasc Pathol. 1993;2:137S–148S. [Google Scholar]
- 2.Salzman E W, Merrill E W, Hent K C. In: Hemostasis and Thrombosis. Colman R W, Hirsh J, Marder V J, Salzman E W, editors. Philadelphia: Lippincott; 1994. pp. 1469–1485. [Google Scholar]
- 3.Milton Harris J, Zalipsky S, editors. Poly(ethylene glycol): Chemistry and Biological Applications. Washington, DC: Am. Chem. Soc.; 1997. [Google Scholar]
- 4.Andrade J D, editor. Surface and Interfacial Aspects Of Biomedical Polymers. New York: Plenum; 1985. [Google Scholar]
- 5.Torchilin V P, Trubetskoy V S. Adv Drug Delivery Rev. 1995;16:141–155. [Google Scholar]
- 6.Needham D, McIntosh T J, Simon S A, Zhelev D. Curr Opin Colloid Interface Sci. 1998;3:511–517. [Google Scholar]
- 7.Napper D H. Polymeric Stabilization of Colloidal Dispersions. New York: Academic; 1983. [Google Scholar]
- 8.Holland N B, Yongxing Q, Ruegsegger M, Marchant R E. Nature (London) 1998;392:799–801. doi: 10.1038/33894. [DOI] [PubMed] [Google Scholar]
- 9.Ikada Y. Adv Polym Sci. 1984;57:104–140. [Google Scholar]
- 10.Jeon S I, Lee J H, Andrade J D, de Gennes P G. J Colloid Interface Sci. 1991;142:149–158. [Google Scholar]
- 11.Szleifer I. Curr Opin Solid State Mater Sci. 1997;2:337–344. [Google Scholar]
- 12.Lasic D, Martin F, editors. Stealth Liposomes. Boca Raton, FL: CRC; 1995. [Google Scholar]
- 13.Fisher L, Lasic D. Curr Opin Colloid Interface Sci. 1998;3:509–510. [Google Scholar]
- 14.McPherson T, Kidane A, Szleifer I, Park K. Langmuir. 1998;14:176–186. [Google Scholar]
- 15.Fraaije J G E M. J Chem Phys. 1993;99:9202–9212. [Google Scholar]
- 16.Hasegawa R, Doi M. Macromolecules. 1997;30:3086–3089. [Google Scholar]
- 17.Kondepudi D, Prigogine I. Modern Thermodynamics: From Heat Engines to Dissipative Structures. New York: Wiley; 1998. [Google Scholar]
- 18.Marconi U M B, Tarazona P. J Chem Phys. 1999;110:8032–8044. [Google Scholar]
- 19.Ravichandran S, Talbot J. Biophys J. 2000;78:110–120. doi: 10.1016/S0006-3495(00)76577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szleifer I, Carignano M A. Adv Chem Phys. 1996;94:165–260. [Google Scholar]
- 21.Szleifer I. Curr Opin Colloid Interface Sci. 1996;1:416–423. [Google Scholar]
- 22.Szleifer I. Biophys J. 1997;72:595–612. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S J, Park K. J Vac Sci Technol. 1994;12:1–7. [Google Scholar]
- 24.Prime K L, Whitesides G M. Science. 1991;252:1164–1167. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- 25.Prime K L, Whitesides G M. J Am Chem Soc. 1993;115:10714–10721. [Google Scholar]
- 26.Halperin A, Tirrell M, Lodge T P. Adv Pol Sci. 1992;100:31–71. [Google Scholar]