

Abstract
The pathophysiologies of osteoporosis, cardiovascular disease, and breast cancer are briefly reviewed within the context of the relevance and safety of long-term estrogen therapy (ET). Extrapolation of data from the known underlying biology of these diseases and the results of randomized controlled clinical trials suggest that selective ET is appropriate and safe for the majority of postmenopausal women. A key element to this clinical practice is individualization of ET, which includes timing of the initiation of therapy, selection of the route and possibly the type of estrogen prescribed, adjustment of the dose of estrogen over time to compensate for local tissue estrogen synthesis, and annual monitoring and reassessment of the indication for continuing therapy. Established disease requires disease-specific therapy but does not exclude ET cotherapy provided there is an indication for its use. Estrogen-dependent cancer is an absolute contraindication to systemic ET.
Readers are encouraged to respond to George Lundberg, MD, Editor of MedGenMed, for the editor's eye only or for possible publication via email: glundberg@medscape.net
Introduction
Estrogen therapy (ET) is pharmacologic but can be adjusted to replicate the physiologic endocrine milieu of women in order to achieve and maintain the physiologic homeostasis of various organ functions. Recognition of the age-related pathogenesis of ET-responsive conditions and diseases is essential (Figure 1). For example, hot flashes frequently precede the menopause but decrease in prevalence and intensity over time. Symptoms associated with urogenital atrophy (UGA) postdate the onset of menopause but increase in frequency and severity with aging. Each of these symptom complexes has the same etiology – estrogen deficiency – but they differ in terms of timing, type, and duration of ET that would be appropriate. Osteoporosis, cardiovascular disease (CVD), and breast cancer have multifactorial etiologies, with endogenous estrogen being a contributing protective or aggravating factor.
Figure 1.
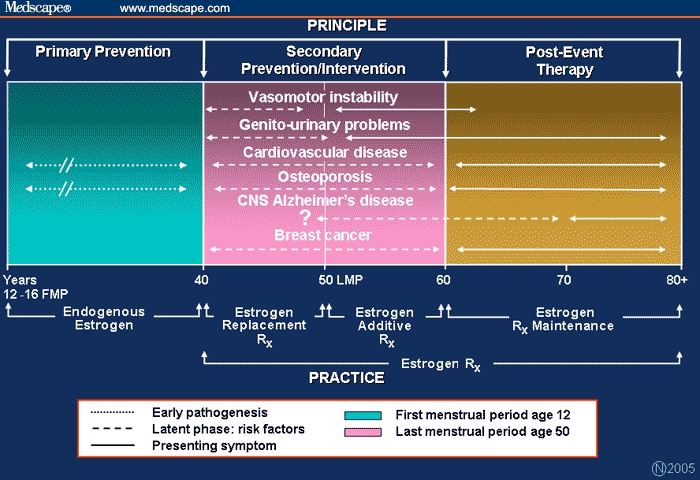
Age-adjusted hormone therapy and the pathogenesis of estrogen-related conditions.
Timing of exogenous ET and matching the dose, route, and possibly the duration of therapy to meet the needs of a given individual require adjustment to ensure appropriate and safe therapy. In this context, there are 3 pharmacologic approaches one might consider, each of which is governed by the pharmacokinetics of the ET and the age and health of the individual (Figure 1). The first is estrogen replacement therapy (ERT), which sets out to replicate the estrogen milieu of premenopausal women (both in terms of the blood levels of estrogen and ratio of estradiol to estrone [E2/E1]). The second is estrogen additive therapy (EAT), which entails complementing endogenous postmenopausal estrogen with exogenous estrogen, tailored to meet the indication for ET. And finally, estrogen maintenance therapy (EMT) is an extension of previously prescribed ET, but at gradually reduced dosages, with the objective of maintaining the individual's state of well-being.
The initiation and type of ET required to meet these goals vary: ERT is relevant to women experiencing a premature or early menopause (ie, < 50 years of age) or premature ovarian failure; EAT is the most common form of ET prescribed, primarily for management of the symptomatic menopause, and when indicated, for the prevention of osteoporosis (50 to 60 years of age); EMT is less commonly practiced (or recommended) and has to be balanced with the potential for estrogen-associated breast cancer (> 60 years of age).
Irrespective of age, appropriate lifestyle, exercise, and nutritional measures should be advised and prescribed. Disease-specific drugs are required for overt conditions, eg, diabetes, dyslipidemia, and hypertension, but these diseases do not necessarily contraindicate the concomitant use of ET.
Osteoporosis
Sufficient bone mass and an intact trabecular microarchitecture are 2 of the main factors responsible for bone strength and prevention of osteoporotic fracture. Prevention of falls is an additional factor that reduces fracture risk.
Biologic Rationale
Achieving peak bone mass and maintaining bone mass depend on the balanced coupling of new bone formation (osteoblast function) and bone removal, or resorption (osteoclast activity) in the bone remodeling cycle. Osteocytes modulate the osteoclast/osteoblast interaction and play a role as ‘mechanotransducers’ in the positive effect of mechanical loading (exercise) on bone strength. The sex steroids are involved in development of peak bone mass and skeletal homeostasis.[1] Estrogens and androgens influence bone homeostasis through effects on osteoblasts and osteoclasts that are mediated by the steroid receptors[2] and through gene regulation (eg, estrogen-regulated genes with trabecular bone-sparing effects have been identified[3]).
Estrogen is synthesized endogenously in bone tissue in part by the aromatization of testosterone to estradiol (E2).[4] Estrogen upregulates steroid receptors,[5] and the estrogen receptors (ERs) are downregulated with aging and estrogen deprivation. Apoptosis of osteocytes is closely linked to estrogen deficiency and may be one reason why mechanical loading does not prevent bone loss in the presence of low E2 levels.[6] Estrogen also has important extracellular functions that influence bone health. There is evidence from animal studies that estrogen regulates genes involved in a vitamin D-independent transport of calcium from the intestines and renal tubules; estrogen increased the levels of activated vitamin D [1.25(OH)2D3] and vitamin D-binding protein.[7] There are specific vitamin D receptors in muscle,[8] and apart from its role in calcium metabolism, vitamin D improves muscle strength and prevents body sway.[9]
Clinical Trials
The literature is replete with randomized controlled clinical trials (RCTs) that have clearly demonstrated a positive effect of standard doses of estrogen on bone mass accrual and improvement in bone mineral density (BMD). In a study of postmenopausal women (> 65 years), those who had undetectable levels of endogenous estrogen (< 5 pg/mL) were found to be at increased risk of hip and vertebral fracture, as were postmenopausal women with higher endogenous serum concentrations of sex hormone binding globulin (SHBG).[10] The risk of fracture decreased with serum estrogen levels above 10 pg/mL but within normal postmenopausal concentrations (< 20 pg/mL), which the authors proposed could be achieved with low-dose ET without significant risk for endometrial or breast cancer.[10]
A series of studies have confirmed that exogenous estrogen can increase BMD in doses much lower than the standard dose of estrogen prescribed for menopausal symptoms (eg, conjugated equine estrogens [CEE], 0.625mg). These include oral micronized E2, 0.25 mg[11]; transdermal E2, 25 mcg[12]; CEE, 0.3 mg and 0.45 mg[13]; and ethinyl estradiol (EE), 5 mcg.[14] The increase in BMD achieved with these lower doses averaged about 1% to 3% over a 2-year study interval – approximately half of that following standard doses of estrogen. However, none of the studies with these low-dose regimens evaluated fracture. More recently, an ultra low-dose regimen of unopposed transdermal E2 (14 mcg) reported a 2% increase in BMD in the lumbar spine of older postmenopausal women (mean age, 67 years) relative to placebo (P < .001) without causing endometrial hyperplasia.[15]
Urinary bone markers (deoxypyridinoline; type I collagen breakdown products) above the upper limit of normal for premenopausal women are predictive of hip fracture. The initial increase and subsequent gradual decrease in postmenopausal bone remodeling may influence clinical decisions regarding the timing and dose of ET. For example, in a study of healthy, recently postmenopausal women, the percentage of positive responders (ie, who had a positive change from baseline in lumbar BMD) on transdermal ET correlated with the ET dose: 59.6%, 25 mcg; 79.3%, 50 mcg; and 83.9%, 75 mcg.[12] A similar result was found for women on low-dose oral E2 therapy.[11] In clinical practice, the nonresponders could be identified by biochemical monitoring and their dose of ET could be titrated upwards to ensure a positive clinical response.
The estrogen-alone arm of the Women's Health Initiative (WHI) reported a 39% reduction in hip fracture and a 38% decrease in vertebral fractures.[16] The results are consistent with earlier observational studies and recent meta-analyses. It also links the biologic plausibility of improvement in BMD with a reduced risk of fracture as reported in other studies involving the use of nonhormonal antiresorptive agents. Notably, the protective effect of ET in the WHI study for hip fractures was confined to women older than 60 years of age, and particularly in the age 60 to 69 category: RR 0.33 (0.13-0.83) vs RR 0.62 (0.38-1.00) in women aged 70 to 79 years. The protective effect of ET on bone declines rapidly upon cessation of treatment.[17]
Clinical Practice
Given the asymptomatic nature of osteoporosis, screening with peripheral or axial dual energy x-ray absorptiometry (DXA) is recommended. Women with osteopenia are candidates for preventive therapy, especially if they have evidence of accelerated bone remodeling. In this context, pretreatment assessment of the biochemical bone markers,[18] especially if the values are above the premenopausal reference range, may be valuable for treatment decision making, selecting an ET dose, and monitoring the efficacy of ET. Reduction of the pretreatment bone turnover markers (eg, urinary collagen cross-linked N telopeptide) by 50% from the baseline value is indicative of biologic efficacy, and some studies, but not all, have found bone markers useful for predicting risk of future fracture.[18–23]
Timing of therapy and the dose of ET are more important than the choice of estrogen. The presence of other indications for ET and the effect of ET on SHBG should also be factored into the decision. On this basis, I would recommend that recently symptomatic menopausal women should be treated with either oral E2 (1 mg), CEE (0.45 mg), or the transdermal E2 patch (50 mcg). As indicated by the WHI study, the protective effect of estrogen occurs beyond the early menopausal period.[16] Guided by annual biochemical marker monitoring and 2-yearly DXA monitoring, the dose of ET may be gradually titrated downwards (eg, every 5 years) until a much lower maintenance (but clinically effective) dose of ET is achieved.
Cardiovascular Health and Disease
Cardiovascular disease (CVD) is the leading cause of death in women. Atherosclerosis, the major underlying pathology, is a multifactorial disease that develops from early childhood but only presents clinically in women a decade or more beyond menopause, as atherothrombosis (myocardial infarction or stroke). The majority of women in Western societies have varying degrees of atherosclerosis as they enter menopause. Central to this issue is the timing of ET.
Biologic Variables - Metabolic Factors
Lipid and Lipoprotein Metabolism
The majority of women (66%) in the Framingham Study who developed incident coronary heart disease (CHD) during a 12-year follow up did not have elevations of their total cholesterol or low-density lipoprotein (LDL)-cholesterol.[24] The most common lipid abnormalities in these women were elevated triglycerides and low high-density lipoprotein (HDL)-cholesterol. Individuals with elevated triglyceride levels have an increased concentration of LDL-cholesterol; the particles are reduced in size and are therefore more atherogenic. Triglyceride values in excess of 200 mg/dL are associated with increased amounts of atherogenic remnant lipoproteins, which may substantially elevate the risk for CVD beyond that predicted on the basis of LDL-cholesterol alone. Hypertriglyceridemia results in decreased HDL-cholesterol and impairment of reverse cholesterol transport, and is also associated with insulin resistance, glucose intolerance, hypertension, and prothrombotic states.[25] High levels of the apolipoprotein Lp(a) have been identified as an independent risk factor for CVD. Concentrations of Lp(a) tend to increase after menopause.[26]
Glucose, Insulin Resistance, and Diabetes
CVD is more prevalent in women with both insulin-dependent and independent diabetes and latent glucose intolerance.[27,28] The association between abnormal carbohydrate metabolism and the pathogenesis of atherosclerosis is unclear: hyperglycemia has an adverse effect on the vessel wall; hyperinsulinemia appears to be an independent risk factor acting through alternative mechanisms.[29] The menopause is associated with a progressive decrease in insulin sensitivity, and insulin resistance is more common in postmenopausal than premenopausal women.[30]
Clinical Message
Hypertriglyceridemia serves as an excellent surrogate marker for dyslipidemia, hypercoagulability, and insulin resistance. Modifying factors related to hypertriglyceridemia may in turn improve insulin sensitivity, fibrinolysis, and, therefore, the potential for CVD.
Blood Pressure and Hypertension
Estrogen vasodilates arterial vessels via a number of mechanisms that have a direct effect on the smooth muscle of the arterial wall. This is also true for estrogen metabolites that may be even more potent than estradiol.[31]
Vascular Endothelium in Health and Disease
Two mechanistic processes influence arterial wall disease and function: stability of the atheromatous plaque and vasoreactivity of the arterial endothelium.
Atheromatous Plaque
Plaque stability is determined by pro-inflammatory cytokines, such as monocyte chemoattractant protein (MCP-1), and cell adhesion molecules that recruit monocytes from the blood into the intima of the arteries where they subsequently become lipid-laden foam cells.[32,33] C-reactive protein (CRP), an acute-phase inflammatory biomarker, has emerged as one of the most important predictors of CVD.[34,35] Mature plaques consist of a lipid-rich core and a fibrous cap of extracellular matrix proteins, and elevated plasminogen activator inhibitor-1 (PAI-1) levels are associated with thin wall plaques.[36] Fissuring of the weakest cap sites are initiated by proteinases that degrade the cap.[37] One such proteinase is matrix metalloproteinase (eg, MMP-9), which accumulates at the edges of atherosclerotic plaques. Estrogen increases circulating levels of MMP-9 in postmenopausal women.[38] Thus, postmenopausal women who have a systemic and/or local predisposition to rupture of atherosclerotic plaques (ie, have high endogenous PAI-1 levels and/or thin plaques) may be at increased risk for an estrogen-associated cardiac event prior to the initiation of ET.
Arterial Vasoreactivity
Estrogen has an essential role in mediating arterial vasodilation and blood flow.[39] This is achieved by stimulating endothelial prostacyclin synthesis (a potent vasodilator) and by inhibiting the vasoconstrictive effect of endothelin-I on the arterial subendothelial smooth muscle cells.[40,41] Estrogen also mediates vascular reactivity via nitric oxide synthesis. The respective roles of ERalpha and ERbeta in this regard are unclear, but animal studies suggest that ERbeta may play a significant role. Atherosclerosis decreases the number of ERs (71% in healthy arteries vs 32% in atherosclerotic arteries) and increases the degree of DNA methylation of ERalpha (which alters its expression).[42,43] The reduced ER expression in atherosclerotic lesions may be further compromised by exogenous estrogen plus progestin therapy (HT) - namely, by the co-prescribed progestin, which has been found in animal studies to reverse estrogen's atheroprotective effects. The bioavailability of the co-prescribed estrogen in combined continuous HT products to compensate for this potential adverse event is, therefore, important.
Clinical Trials
A meta-analysis of 248 studies reviewed the effects of 42 HT regimens on lipid and lipoprotein concentrations.[44] All estrogen-alone regimens raised HDL-cholesterol levels and lowered LDL and total cholesterol levels. The results of this meta-analysis confirmed more recent studies finding that EE 5 mcg increased triglycerides by approximately 39% over baseline[14]; the comparable mean triglyceride elevations for CEE (0.625 mg) and E2 (1 mg) were 35% and 10%, respectively.[45,46]
Oral ET has been shown to have a bimodal effect on insulin sensitivity and glucose tolerance.[47] Standard-dose CEE (0.625 mg) improved insulin sensitivity; higher doses of CEE (1.25 mg) resulted in a deterioration of insulin activity.[47] Other studies have shown that transdermal ET improves or does not adversely influence insulin activity.[48–50] Improvement in insulin sensitivity with an estrogen-alone or an estrogen plus progestin regimen has been reported in both nondiabetic postmenopausal women[49,51–54] and postmenopausal women with type 2 diabetes.[55–57] Other studies have found that insulin sensitivity is unaffected or reduced with estrogen-alone or estrogen plus progestin regimens.[49–51,58–61] There is an interindividual variation in response to glucose tolerance among women on ET: subjects with higher basal fasting glucose or insulin levels appear to have an improved insulin sensitivity and clinical response to ET, suggesting that ET may have the ability to pharmacologically restore glucose homeostasis to normal.[62] This is consistent with results from the WHI that showed a reduced risk of non-insulin-dependent diabetes in current hormone therapy users.[63,64]
Although estrogen lowers blood pressure, some women have an idiosyncratic response to ET with elevation in blood pressure. This response occurs most often with oral ET. It is associated with an increased production of renin substrate by the liver and a reversible stimulation of the renin-angiotensin-aldosterone system,[31] and is more likely to occur with CEE due to abnormal hepatic protein synthesis and nonphysiologic angiotensin I/II cleavage products.[65] Oral or transdermal E2 is the preferred estrogen to prescribe under these circumstances.[31] Hypertension should generally not be a contraindication to ET.[31]
The authors of the Estrogen in the Prevention of Atherosclerosis (EPAT) study in healthy postmenopausal women with subclinical atherosclerosis reported that 1 mg of oral E2 slowed the progression of atherosclerosis compared with matched placebo-treated women.[66] This finding was more pronounced in women who were not taking lipid-lowering drugs. This result differs from those of an earlier study, the Estrogen Replacement on Progression of Coronary Artery Atherosclerosis (ERA) trial, in which CEE failed to slow the progression of coronary artery atherosclerosis in women with established disease.[67] The main differences between these studies were younger age and absence of overt disease in the women studied in EPAT. Further RCTs will be needed to determine whether other estrogens have the same positive effect on subclinical atherosclerosis as unopposed E2.
E2 and CEE improve brachial artery flow-mediated dilation, a surrogate biomarker of coronary artery vascular reactivity.[41] Treatment of postmenopausal women with a variety of ETs reduces the pro-inflammatory cytokine MCP-I concentration to premenopausal levels. The same was found for cell adhesion molecules, all of which were reduced with ET.[68] CEE has been reported in to increase CRP, but the observed associated increase in cardiac events was related to baseline pretreatment CRP concentrations.[69] However, a recent analysis has found that it was progestin in combination with CEE that potentiated the IL-6 (inflammatory)-mediated stimulation of CRP, and not CEE alone.[70] Transdermal E2 is less likely to stimulate an increase in CRP, and a recent review found that whereas oral hormone therapy was generally associated with elevated CRP transdermal E2, it was not associated with elevated levels of IL-6 or CRP alone or with the addition of progestins.[71]
The use of CEE alone therapy in the WHI study was not associated with an increase in the incidence of CHD when the data for the total trial group were analyzed – hazard ratio (HR) = 0.91 (0.73-1.14).[16] The HR for the younger subjects (aged 50 to 59 years), however, was reduced (HR 0.56 [0.3-1.03]), although the confidence limits in this category just exceeded statistical significance. This result is nevertheless consistent with the 50% reduction in CVD noted in the majority of observational studies. Younger age and earlier initiation of ET may be reflective of a healthy user effect that is relevant to the majority of women who seek treatment for symptoms during their early menopause. The effect of lower-dose ET (CEE 0.45 mg and equivalent doses with other estrogens) on cardiovascular health is not known. Lower-dose ET seems to have a similar protective effect as standard-dose ET on metabolic risk factors associated with CVD. [72,73]
Clinical Practice
Animal experiments provide validated and biologically sound data documenting the ability to prevent cardiovascular disease with early ET. However, application of these data to clinical practice is not advocated due primarily to the absence of comparable data from long-term RCTs in younger healthy women. Subanalysis of data from studies reporting adverse cardiovascular events frequently note that a significant percentage of these subjects had an underlying latent abnormality that would have precluded ET. In short, it is often the inappropriate selection of a patient for ET that is responsible for undesired side effects, rather than the ET per se.
Screening of patients before initiating treatment for any estrogen-related indication should include a metabolic profile (eg, glucose, liver enzymes, LDL and HDL cholesterol, triglycerides, and CRP). The choice of the type of estrogen and the route of ET will be governed by the result. The lowest dose of ET consistent with the indication should be prescribed, and the patient needs to be monitored regularly to assess the efficacy of treatment. All available ETs are safe for the majority of eligible menopausal women. The presence of a metabolic abnormality (eg, dyslipidemia, glucose intolerance, or hypertension) is not a contraindication for ET. The type, dose, and sometimes the route of ET may need to be adjusted according to the pharmacologic profile of the ET. Specific cardiovascular diseases may need treatment with disease-specific drugs.
Hemostasis and Venous Thrombosis
Venous thrombosis (deep vein thrombosis [DVT] and pulmonary embolism [PE]) is a relatively uncommon condition.[74,75] The pathogenesis of venous thrombosis differs from that of arterial thrombosis. Venous thrombosis is associated with a strong genetic predisposition to hypercoagulation and a contributory decrease in venous blood flow. Arterial thrombosis results from the stimulation of platelets and the initiation of the coagulation cascade, secondary to a damaged vascular endothelium. The commonality between venous and arterial thrombosis is formation of a thrombus. Risk factors and the clinical profile for VTE and arterial disease differ.[74]
Biology: Coagulation
Hemostasis is a delicately balanced process involving a coagulation cascade that has an intrinsic and an extrinsic pathway.[74] A number of inactive zymogens (factors) are converted to proteases (activated factors) that eventually result in the conversion of prothrombin to thrombin. Thrombin catalyzes fibrinogen to fibrin and leads to the formation of an insoluble blood clot. The coagulation cascade is modulated by an anticoagulation system involving primarily antithrombin III (a factor responsible for 50% of the fluidity of blood), protein C, and its cofactor protein S. Fibrin formation is controlled by the fibrinolytic activity of plasmin, which in turn is derived from the activation of the zymogen, plasminogen. This system is controlled by activators (plasminogen activator [t-PA]) and inhibitors, PA1-1.
Platelets have a central role in arterial thrombosis. They adhere to various adhesion molecules and to the exposed subendothelium of the damaged arterial wall. Platelet activation mediates coagulation via the intrinsic pathway, to factor X and thrombin formation. Venous thrombosis is initiated via the extrinsic pathway.
Clinical Trials
The type, dose, and route of ET are important in terms of venous thrombosis. Oral esterified estrogen has been shown not to be associated with an increase in VTE risk (odds ratio [OR] 0.92 [0.69-1.22]) compared with an equivalent dose of CEE (OR 1.65 [1.24-2.19]).[76] A range (0.9 to 2.5 mg) of CEE doses were studied, and there was greater difference with higher daily doses of CEE. The OR was 1.21 [0.46-3.17] with low-dose CEE (mean, 0.3 mg) and 3.80 [1.90-7.61] with high-dose (mean 1.16 mg) CEE.[76]
Since the procoagulant factors are synthesized in the liver, avoidance of the entero-hepatic first-pass effect is associated with a more limited change in the hemostatic profile.[77] Whether this is clinically important needs to be investigated. Most of the changes in the biomarkers of coagulation, anticoagulation, and fibrinolysis result from oral estrogen usage (CEE, E2, or EE). These include an increase in the levels of tissue activator fibrinolysis inhibitor antigen, protein C, D-dimer, and factors VII, IX, and X (all indicators of increased coagulation), and a decrease in levels of protein S, antithrombin III (anticoagulation), and tissue plasminogen activators (fibrinolysis).[78] The degree to which ET increases the risk of VTE will vary primarily with the dose of estrogen and possibly the route of ET. In one study, the OR for VTE in current users of oral and transdermal ET compared with nonusers was 3.5 (1.8-6.8) and 0.9 (0.5-1.6), respectively.[77] The estimated risk for VTE in current users of oral ET compared with transdermal ET users was 4.0 (1.9-8.3).
The risk of VTE (including DVT and PE) in the estrogen-alone (CEE 0.625 mg) arm of the WHI was 28 (treatment group) vs 21 (placebo group) per 10,000 person-years,[16] a finding consistent with earlier data from observational studies. These results must be judged in the context of (1) the relative infrequency of VTE in adults (1 or 2 per 1000) and the 2- to 4-fold increase associated with ET; (2) age; the risk is significantly lower in younger women; (3) weight; a similar increase in the risk of VTE is related to body mass index (BMI), and (4) genetic predisposition; women on ET with heterogeneous and homogenous Factor V Leiden disease polymorphism, for example, have a significant 7.5- to 8-fold increased risk for VTE.[79]
In 2 recent studies in healthy postmenopausal women, CEE (0.625 mg) significantly decreased levels of anticoagulation factors (fibrinogen, antithrombin III, total and free protein S, and t-PA and PAI-1) and increased coagulation factors (VII and D-dimer).[80,81] However, a study that looked at lifestyle factors found that BMI, high levels of total cholesterol and tricglycerides, and low HDL levels had more of an impact on hemostatic variables in healthy postmenopausal women than hormone therapy or menopausal status.[82]
Clinical Practice
Venous thrombosis is primarily a balance between coagulation and anticoagulation. Interpretation of ET-related changes in the relevant factors must be applied with due caution in practice for the following reasons. Most of the factors assayed are in their inactive form (zymogens), and the system has a built-in redundancy. For example, antithrombin III activity has to be reduced by 50% before the disease is clinically relevant. In addition, significant changes in coagulation may be balanced by an equivalent alteration in anticoagulation.[78] There are no tests appropriate for routine clinical use that assay platelet activity.[77,78,83] On the basis of available data and clinical experience, ET has no clinically significant adverse effects on coagulation in healthy postmenopausal women.
The Breast and Breast Cancer
Fear of breast cancer is the primary reason why many menopausal women elect not to take hormone therapy. The common perception is that the disease is caused by estrogen despite contrary evidence. The single greatest risk factor for breast cancer is aging; the highest prevalence of this disease occurs at a time when systemic estrogen levels are the lowest.[84]
Biology: Breast Cancer Is Not a Single Disease
Although linked by a common histology, breast cancer is a varied and complex multifactorial disease. Estrogen does have an important role in its development, but there is much evidence to support the concept that exogenous estrogen (ie, ET) may serve as a promoter and not an initiator of breast cancer. Central to this issue is the genetic polymorphism of younger women (< 50 years) who may have gene mutations controlling breast cell cycle growth (mutated BRCA1, BRCA2, P53 genes) vs postmenopausal women (> 50 years) with a genetic predisposition to increased or aberrant breast tissue estrogen synthesis and metabolism.[85] Estrogen-associated breast cancer is more prevalent in women with a predominant ERalpha/ERbeta distribution.[86]
Women with breast cancer have higher tissue levels of estrogen. This has been correlated with increased enzymatic activity (aromatase, sulfatase, and 17beta OH dehydrogenase).[87] The synthesis of estrogen takes place in the stromal tissue of the breast. Estrogen-dependent growth factors are stimulated via paracrine signaling and induce specific pathways that effect cell cycle genes.[88] ERalpha and ERbeta are found in the ductal epithelium. ERalpha is co-localized with the progestin receptor (PR), of which there are 2 isoforms. PRB is the active receptor; PRA is either inactive or acts as an inhibitor of PRB. Approximately 10% to 20% of epithelial cells are ERalpha and PR positive and are distributed in both the intralobular duct and the peripheral alveoli.[88]
Estrogen-related breast cancer is preceded histologically by atypical epithelial hyperplasia.[89] The site for the transformation of normal to abnormal cell growth is the terminal duct lobuloalveolar unit (TDLU).[90] The degree of differentiation of the TDLUs determines the percentage of ERalpha and PR-positive cells.[88] The percentage of steroid receptor-positive cells decreases progressively as the lobules mature. Factors influencing this include the duration of premenopausal exposure to estrogen and pregnancy-induced differentiation and maturation of the ductal epithelium. Thus, women who have an early menarche and a late menopause are at increased risk of breast cancer; early and repeated pregnancy reduces the risk of breast cancer.[90]
In short, women who develop breast cancer while taking ET or HT are probably genetically programmed to modify physiologic pathways in response to hormonal therapy and/or environmental carcinogens.
Clinical Trials
Mammographic density reflects the breast's hormonal environment, the influence of background genetics regulating this environment, and the effect of various exogenous hormone therapies and other therapies.[91] Approximately 25% of HT users have dense breasts. Most of the increase in breast density occurs within the first year of treatment and diminishes within 3 weeks of stopping HT.[92] The literature on the association between ET/HT and breast density is extensive and has recently been reviewed.[93–95] Continuous combined HT preparations are more likely to cause an increase in density (20% to 35%) than in estrogen-alone treated women (10% to 20%), and oral ET more so than transdermal ET.[93] The type and dosage of estrogen and co-prescribed progestins are important.[94] Studies of free E1 and E2 in breast adipocytes obtained from postmenopausal women before and after treatment with CEE showed a greater amount of E1 and E2 in the adipocytes both before and after treatment. Pre- and post-ET adipocyte E2 concentrations were 13 pg/mL and 35 pg/mL; the respective estrone values were 209 pg/mL and 626 pg/mL.[96] Relatively low serum estradiol values (> 10 pmol/L [2.7 pg/mL]) are associated with almost a 7-fold higher rate of breast cancer than that of women with undetectable estradiol levels.[97]
The estrogen plus progestin arm of the WHI study confirmed that HT increased the risk of breast cancer (1.26 [1.00-1.59]),[98] a result that is consistent with most of the observational studies and meta-analyses comparing the risk of breast cancer and HT in users vs nonusers.[84,99] The risk of breast cancer increases with the duration of therapy.[84] A minority of women from the estrogen plus progestin arm of the WHI study were adversely affected: 35 vs 30 events annually of 10,000 women on HT and placebo, respectively.[98]
What predisposes the few extra women on relatively short-term ET to develop breast cancer? Apart from the individual's genetically determined biology (and occasional mutations of genes that result in extremely high aromatase activity, for example),[100] progestins may potentially increase the risk of breast cancer. Thus, the recent WHI estrogen-alone study reported a 23% lower rate of breast cancer in women treated with CEE vs placebo (26 vs 33 per 10,000 women-years). The HR of 0.77 narrowly missed statistical significance (P = .06).[16] The subject of progestin-additive therapy is beyond the scope of this review. However, studies in cynomolgus monkeys have shown that CEE and MPA[101] induce significant epithelial proliferation in the mammary glands of castrated monkeys, whereas a combination of EE and NETA does not.[102] This finding is consistent with the results of another simian study, which found that CEE treatment increased the total amount of PR and that the addition of MPA decreased repressor PRA but further increased the proliferating PRB levels. MPA-alone therapy had no effect on the amount of PRA or PRB.[103]
Clinical Practice
The biology of breast tissue and its oncogenic potential cannot be clinically evaluated and monitored. Given the variability of endogenous and exogenous estrogen metabolism, it is prudent to select a dose and route of ET that most closely replicates the age-matched hormonal milieu of the women being treated. Low-dose oral or transdermal E2 has a pharmacologic profile that most closely meets this objective. Mammographic density testing is a valuable surrogate measure of breast tissue estrogenicity.[91]
Global Index and Individualizing ET
The global index is a recently introduced statistical evaluation of population RCTs. Its validation and its applicability to individualized clinical care have not been established. The primary outcome of the estrogen-alone WHI study was CHD incidence, and invasive breast cancer was the primary safety outcome.[16] The results for women aged 50 to 59 years (the candidates most likely to be treated with ET in everyday clinical practice) showed a CHD protective effect (HR 0.56 [0.30-1.03]) and a reduction in breast cancer incidence (HR 0.72 [0.43-1.41]). The colorectal cancer incidence was also reduced, but not hip fracture, although ET in older cohorts (who are at greater risk for hip fracture) was significantly decreased: 60 to 69 years (HR 0.33 [0.18-0.83]); 70 to 79 years (HR 0.62 [0.38-1.00]).[16] Despite this encouraging outcome, the study authors concluded that “[a]t present, these data demonstrate no overall benefit of CEE for chronic disease prevention in postmenopausal women and thus argue against its use in this setting.” This decision was based on the global index of the total group and factored in the risks and benefits of ET, including the primary outcomes plus stroke, pulmonary embolism, colorectal cancer, and hip fracture. The excess risk for all monitored events in the global index was a nonsignificant 2 events per 10,000 person-years. Although there were 12 additional strokes per 10,000 person-years, there were the same number of strokes (19) in both the CEE- and placebo-treated 50- to 59-year-olds. The statement that ET “does not significantly affect the incidence of CHD (the primary outcome)…”[16] after an average of 6.8 years of ET may be applicable to women in the older groups. Given the natural history of atherothrombotic disease, however, the women in the 50 to 59 year age range had not yet reached the chronologic age at which women typically experience myocardial infarction and related CHD events. Extension of the study for 5 or more years may have decided this issue.
In practice, an individualized global (index) evaluation is (or should be) performed on every patient before prescribing hormone or any other pharmacologic therapy. Thorough clinical evaluation of the total healthcare needs of every woman and an understanding of the underlying biology of a given condition are central to appropriate and individualized pharmacologic intervention. Results of clinical trials do provide guidance and direction. For example, the Multiple Outcomes of Raloxifene Evaluation (MORE) trial[104] found women without prevalent fractures had a lower risk for future fractures and cardiovascular events but a greater risk for breast cancer than matched cohorts with a prevalent fracture on study entry. This result dovetails with the known biology and hormonal contribution to these diseases: higher endogenous E2 levels are associated with an increased risk of breast cancer but a lower risk of osteoporosis (and vice versa). It is appropriate to consider all relevant scientific data and to base clinical decisions on the evidence. However, this should also include validated biologic evidence, which is the foundation for the 3 basic sciences on which medical training and practice are founded: physiology, pathology, and pharmacology. The biologic heterogeneity in the pathogenesis of all conditions and diseases previously discussed is clear. The pharmacologic differences in available ETs are well documented. The resulting variables and reality of clinical practice confirm that individualized ET can only be achieved by personalized evaluation and pharmacologic intervention.
Funding Information
This article was supported in part by an independent and unrestricted educational grant from Novo Nordisk.
References
- 1.Lindberg MK, Vandenput L, Moverare Skrtic S, et al. Androgens and the skeleton. Minerva Endocrinol. 2005;30:15–25. [PubMed] [Google Scholar]
- 2.Balasch J. Sex steroids and bone: current perspectives. Hum Reprod Update. 2003;9:207–222. doi: 10.1093/humupd/dmg017. [DOI] [PubMed] [Google Scholar]
- 3.Lindberg MK, Moverare S, Eriksson A-L, et al. Identification of estrogen-regulated genes of potential importance for the regulation of trabecular bone density. J Bone Miner Res. 2002;17:2183–2195. doi: 10.1359/jbmr.2002.17.12.2183. [DOI] [PubMed] [Google Scholar]
- 4.Notelovitz M. Androgen effects on bone and muscle. Fertil Steril. 2002;77(suppl 4):S34–S41. doi: 10.1016/s0015-0282(02)02968-0. [DOI] [PubMed] [Google Scholar]
- 5.Rickard DJ, Waters KM, Ruesink TJ, et al. Estrogen receptor isoform-specific induction of progesterone receptors in human osteoblasts. J Bone Miner Res. 2002;17:580–592. doi: 10.1359/jbmr.2002.17.4.580. [DOI] [PubMed] [Google Scholar]
- 6.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–67. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 7.Christakos S, Prince R. Estrogen, vitamin D and calcium transport. J Bone Miner Res. 2003;18:1737–1739. doi: 10.1359/jbmr.2003.18.10.1737. [DOI] [PubMed] [Google Scholar]
- 8.Bischoff-Ferrari HA, Borchers M, Gudat F, et al. Vitamin D receptor expression in human muscle tissue decreases with age. J Bone Miner Res. 2004;19:265–269. doi: 10.1359/jbmr.2004.19.2.265. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer M, Begerow B, Minne HW, et al. Vitamin D status, trunk muscle strength, body sway, falls, and fractures among 237 postmenopausal women with osteoporosis. Exp Clin Endocrinol Diabetes. 2001;109:87–92. doi: 10.1055/s-2001-14831. [DOI] [PubMed] [Google Scholar]
- 10.Cummings SR, Browner WS, Bauer D, et al. Endogenous hormones and the risk of hip and vertebral fractures among older women. N Engl J Med. 1998;339:733–738. doi: 10.1056/NEJM199809103391104. [DOI] [PubMed] [Google Scholar]
- 11.Prestwood KM, Kenney AM, Kleppinger A, Kulldorf M. Ultralow-dose micronized 17beta estradiol and bone density and bone metabolism in older women. A randomized control trial. JAMA. 2003;290:1042–1048. doi: 10.1001/jama.290.8.1042. [DOI] [PubMed] [Google Scholar]
- 12.Notelovitz M, John VA, Good WB. Effectiveness of Alora estradiol matrix transdermal delivery system in improving lumbar bone mineral density in healthy postmenopausal women. Menopause. 2002;9:343–353. doi: 10.1097/00042192-200209000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Lindsay R, Gallagher IC, Kleerekoper M, Pickar JH. Effect of lower doses of conjugated equine estrogen with and without medroxyprogesterone acetate, on bone in early postmenopausal women. JAMA. 2002;287:2668–2676. doi: 10.1001/jama.287.20.2668. [DOI] [PubMed] [Google Scholar]
- 14.Speroff L, Rowan J, Symons J, et al. The comparative effect on bone density, endometrium and lipids of continuous hormones as replacement therapy (CHART study). A randomized controlled trial. JAMA. 1996;276:1397–1403. [PubMed] [Google Scholar]
- 15.Ettinger B, Ensrud KE, Wallace R, et al. Effects of ultralow-dose transdermal estradiol on bone mineral density: a randomized clinical trial. Obstet Gynecol. 2004;104:443–451. doi: 10.1097/01.AOG.0000137833.43248.79. [DOI] [PubMed] [Google Scholar]
- 16.Women's Health Initiative Steering Committee. Effects of conjugated equine estrogen on postmenopausal women with hysterectomy. The Women's Health Initiative randomized clinical trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 17.Wasnich RD, Bagger YZ, Hosking DJ, et al. Early Postmenopausal Intervention Cohort Study Group. Changes in bone density and turnover after alendronate or estrogen withdrawal. Menopause. 2004;11(6 Pt 1):622–630. doi: 10.1097/01.gme.0000123641.76105.b5. [DOI] [PubMed] [Google Scholar]
- 18.Garnero P, Sornay-Rendu E, Duboeuf F, Delmas PD. Markers of bone turnover predict postmenopausal forearm bone loss over 4 years: the Offely study. J Bone Miner Res. 1999;14:1614–1621. doi: 10.1359/jbmr.1999.14.9.1614. [DOI] [PubMed] [Google Scholar]
- 19.Cosman F, Nieves J, Woelfert L, et al. Parathyroid hormone added to established hormone therapy: effects on vertebral fracture and maintenance of bone mass after parathyroid hormone withdrawal. J Bone Miner Res. 2001;16:925–931. doi: 10.1359/jbmr.2001.16.5.925. [DOI] [PubMed] [Google Scholar]
- 20.Lofman O, Magnusson P, Toss G, Larsson L. Common biochemical markers of bone turnover predict future bone loss: a 5-year follow-up study. Clin Chim Acta. 2005;356:67–75. doi: 10.1016/j.cccn.2004.12.014. Epub 2005 Mar 17. [DOI] [PubMed] [Google Scholar]
- 21.Gluer MG, Minne HW, Gluer CC, et al. Prospective identification of postmenopausal osteoporotic women at high vertebral fracture risk by radiography, bone densitometry, quantitative ultrasound, and laboratory findings: results from the PIOS study. J Clin Densitom. 2005;8:386–395. doi: 10.1385/jcd:8:4:386. [DOI] [PubMed] [Google Scholar]
- 22.Briot K, Roux C. What is the role of DXA, QUS and bone markers in fracture prediction, treatment allocation and monitoring? Best Pract Res Clin Rheumatol. 2005;19:951–964. doi: 10.1016/j.berh.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 23.Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin. 2005;21:545–554. doi: 10.1185/030079905x41390. [DOI] [PubMed] [Google Scholar]
- 24.Lloyd-Jones DM, O'Donnell CJ, D'Agostino RB, et al. Applicability of cholesterol lowering primary prevention trials to a general population: the Framingham study. Arch Intern Med. 2001;161:949–954. doi: 10.1001/archinte.161.7.949. [DOI] [PubMed] [Google Scholar]
- 25.Third Report of the National Cholesterol Education Program (NCEP). Detection, evaluation and treatment of high blood cholesterol in adults. (Adult Treatment Panel III) Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 26.Shlipak MG, Simon JA, Vittinghoff E, et al. Estrogen and progestin, lipoprotein(a) and the risk of recurrent coronary heart disease events after menopause. JAMA. 2000;283:1845–1852. doi: 10.1001/jama.283.14.1845. [DOI] [PubMed] [Google Scholar]
- 27.Barrett-Connor E, Giardina EG, Gitt AK, et al. Women and heart disease: the role of diabetes and hyperglycemia. Arch Intern Med. 2004;164:934–942. doi: 10.1001/archinte.164.9.934. [DOI] [PubMed] [Google Scholar]
- 28.Haffner SM. Impaired glucose tolerance, insulin resistance and cardiovascular disease. Diabet Med. (Suppl 3):S12–8. doi: 10.1002/(sici)1096-9136(199708)14:3+<s12::aid-dia439>3.3.co;2-g. 1997 Aug;14. [DOI] [PubMed] [Google Scholar]
- 29.Meigs JB, Mittelman MA, Nathan DM, et al. Hyperinsulinemia, hyperglycemia and impaired hemostasis. The Framingham Offspring Study. JAMA. 2000;283:221–228. doi: 10.1001/jama.283.2.221. [DOI] [PubMed] [Google Scholar]
- 30.Louet JF, LeMay C, Mauvais-Jarvis F. Antidiabetic actions of estrogen: insight from human and genetic mouse models. Curr Atheroscler Rep. 2004;6:180–185. doi: 10.1007/s11883-004-0030-9. [DOI] [PubMed] [Google Scholar]
- 31.Mueck AO, Seeger H. Effect of hormone therapy on BP in normotensive and hypertensive postmenopausal women. Maturitas. 2004;49:189–203. doi: 10.1016/j.maturitas.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Stork S, Bauman K, von Schacky C, et al. The effect of 17beta estradiol on MCP-1 serum levels in postmenopausal women. Cardiovasc Res. 2002;53:642–649. doi: 10.1016/s0008-6363(01)00461-8. [DOI] [PubMed] [Google Scholar]
- 33.Husain T, Abbott CR, Scott DJ, Gough MJ. Macrophase accumulation within the cap of carotid atherosclerotic plaques is associated with the onset of cerebral ischemic events. J Vasc Surg. 1999;30:269–276. doi: 10.1016/s0741-5214(99)70137-0. [DOI] [PubMed] [Google Scholar]
- 34.Labarrere CA, Zaloga GP. C-reactive protein: from innocent bystander to pivotal mediator of atherosclerosis. Am J Med. 2004;117:499–507. doi: 10.1016/j.amjmed.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 35.Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat Clin Pract Cardiovasc Med. 2005;2:29–36. doi: 10.1038/ncpcardio0074. quiz 58. [DOI] [PubMed] [Google Scholar]
- 36.Kohler HP, Grant PJ. Plasminogen activator inhibitor type I and coronary artery disease. N Engl J Med. 2000;342:1792–1801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 37.Yeghiazarians Y, Braunstein JB, Askari A, Stone PH. Unstable angina pectoris. N Engl J Med. 2000;342:101–114. doi: 10.1056/NEJM200001133420207. [DOI] [PubMed] [Google Scholar]
- 38.Zanger D, Yang BK, Ardans, et al. Divergent effects of hormone therapy on markers of inflammation in postmenopausal women with coronary artery disease on appropriate medical management. J Am Coll Cardiol. 2000;36:1797–1802. doi: 10.1016/s0735-1097(00)00952-9. [DOI] [PubMed] [Google Scholar]
- 39.Mendelsohn ME. Protective effects of estrogen on the cardiovascular system. Am J Cardiol. 2002;89:12E–17E. doi: 10.1016/s0002-9149(02)02405-0. discussion 17E-18E. [DOI] [PubMed] [Google Scholar]
- 40.Collins P. Vascular effect of hormones. Maturitas. 2001;38:45–51. doi: 10.1016/s0378-5122(00)00197-3. [DOI] [PubMed] [Google Scholar]
- 41.Williams JK, Delansorne R, Paris J. Estrogens, progestins and coronary artery reactivity in atherosclerotic monkeys. J Steroid Biochem Molec Biol. 1998;65:219–224. doi: 10.1016/s0960-0760(98)00020-x. [DOI] [PubMed] [Google Scholar]
- 42.Losordo DW, Kearney M, Kim EA, et al. Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation. 1994;89:1501–1510. doi: 10.1161/01.cir.89.4.1501. [DOI] [PubMed] [Google Scholar]
- 43.Post WS, Goldschmidt-Clemont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene in association with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43:985–991. doi: 10.1016/s0008-6363(99)00153-4. [DOI] [PubMed] [Google Scholar]
- 44.Godsland IF. Effects of postmenopausal hormone replacement therapy on lipid, lipoprotein and apolipoprotein(a) concentrations: analysis of studies published from 1974-2000. Fertil Steril. 2001;75:898–915. doi: 10.1016/s0015-0282(01)01699-5. [DOI] [PubMed] [Google Scholar]
- 45.Lobo RA, Bush T, Carr BR, Pickar JH. Effects of lower doses of conjugated equine estrogens and medroxyprogesterone acetate on plasma lipids and lipoproteins, coagulation factors and carbohydrate metabolism. Fertil Steril. 2001;76:13–24. doi: 10.1016/s0015-0282(01)01829-5. [DOI] [PubMed] [Google Scholar]
- 46.Davidson MH, Maki KC, Marx P, et al. Effects of continuous estrogen and estrogen-progestin replacement regimens on cardiovascular risk markers in postmenopausal women. Arch Intern Med. 2000;160:3315–3325. doi: 10.1001/archinte.160.21.3315. [DOI] [PubMed] [Google Scholar]
- 47.Lindheim SR, Vijod MA, Presser SC, et al. A possible bimodal effect of estrogen on insulin sensitivity in postmenopausal women and the attenuating effect of adding progestin. Fertil Steril. 1993;60:664–667. doi: 10.1016/s0015-0282(16)56218-9. [DOI] [PubMed] [Google Scholar]
- 48.Godsland IF, Gangar K, Walton C, et al. Insulin resistance, secretion and elimination in postmenopausal women with oral and transdermal hormone replacement therapy. Metabolism. 1993;42:846–853. doi: 10.1016/0026-0495(93)90058-v. [DOI] [PubMed] [Google Scholar]
- 49.Os I, Os A, Abdelnoor M, Larsen A, Birkeland K, Westheim A. Insulin sensitivity in women with coronary heart disease during hormone replacement therapy. J Womens Health (Larchmt) 2005;14:137–145. doi: 10.1089/jwh.2005.14.137. [DOI] [PubMed] [Google Scholar]
- 50.Araujo DA, Farias ML, Andrade AT. Effects of transdermal and oral estrogen replacement on lipids and glucose metabolism in postmenopausal women with type 2 diabetes mellitus. Climacteric. 2002;5:286–292. [PubMed] [Google Scholar]
- 51.Dansuk R, Unal O, Karsidag YK, Turan C. Evaluation of the effects of various gestagens on insulin sensitivity, using homeostatic model assessment, in postmenopausal women on hormone replacement therapy. Gynecol Endocrinol. 2005;20:1–5. doi: 10.1080/09513590400020880. [DOI] [PubMed] [Google Scholar]
- 52.Karim R, Mack WJ, Lobo RA, et al. Determinants of the effect of estrogen on the progression of subclinical atherosclerosis: Estrogen in the Prevention of Atherosclerosis Trial. Menopause. 2005;12:366–373. doi: 10.1097/01.GME.0000153934.76086.A4. Epub 2005 Jul 21. [DOI] [PubMed] [Google Scholar]
- 53.Christodoulakos G, Lambrinoudaki I, Panoulis C, et al. Serum androgen levels and insulin resistance in postmenopausal women: association with hormone therapy, tibolone and raloxifene. Maturitas. 2005;50:321–330. doi: 10.1016/j.maturitas.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Lee CC, Kasa-Vubu JZ, Supiano MA. Differential effects of raloxifene and estrogen on insulin sensitivity in postmenopausal women. J Am Geriatr Soc. 2003;51:683–688. doi: 10.1034/j.1600-0579.2003.00214.x. [DOI] [PubMed] [Google Scholar]
- 55.Borissova AM, Tankova T, Kamenova P, et al. Effect of hormone replacement therapy on insulin secretion and insulin sensitivity in postmenopausal diabetic women. Gynecol Endocrinol. 2002;16:67–74. [PubMed] [Google Scholar]
- 56.Brussaard HE, Gevers Leuven JA, Frolich M, Kluft C, Krans HM. Short-term oestrogen replacement therapy improves insulin resistance, lipids and fibrinolysis in postmenopausal women with NIDDM. Diabetologia. 1997;40:843–849. doi: 10.1007/s001250050758. [DOI] [PubMed] [Google Scholar]
- 57.Andersson B, Mattsson LA, Hahn L, et al. Estrogen replacement therapy decreases hyperandrogenicity and improves glucose homeostasis and plasma lipids in postmenopausal women with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1997;82:638–643. doi: 10.1210/jcem.82.2.3746. [DOI] [PubMed] [Google Scholar]
- 58.Goodrow GJ, L'Hommedieu GD, Gannon B, Sites CK. Predictors of worsening insulin sensitivity in postmenopausal women. Am J Obstet Gynecol. 2006;194:355–361. doi: 10.1016/j.ajog.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 59.Godsland IF, Manassiev NA, Felton CV, et al. Effects of low and high dose oestradiol and dydrogesterone therapy on insulin and lipoprotein metabolism in healthy postmenopausal women. Clin Endocrinol (Oxf) 2004;60:541–549. doi: 10.1111/j.1365-2265.2004.02017.x. [DOI] [PubMed] [Google Scholar]
- 60.Sites CK, L'Hommedieu GD, Toth MJ, Brochu M, Cooper BC, Fairhurst PA. The effect of hormone replacement therapy on body composition, body fat distribution, and insulin sensitivity in menopausal women: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab. 2005;90:2701–2707. doi: 10.1210/jc.2004-1479. Epub 2005 Feb 1. [DOI] [PubMed] [Google Scholar]
- 61.Shadoan MK, Zhang L, Wagner JD. Effects of hormone therapy on insulin signaling proteins in skeletal muscle of cynomolgus monkeys. Steroids. 2004;69:313–318. doi: 10.1016/j.steroids.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 62.Li C, Samsioe G, Borgfeldt C, et al. Low-dose hormone therapy and carbohydrate metabolism. Fertil Steril. 2003;79:550–555. doi: 10.1016/s0015-0282(02)04762-3. [DOI] [PubMed] [Google Scholar]
- 63.Margolis KL, Bonds DE, Rodabough RJ, et al. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women's Health Initiative hormone trial. Diabetologia. 2004;47:1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- 64.Bonds DE, Lasser N, Qi L, Brzyski R, et al. The effect of conjugated equine oestrogen on diabetes incidence: the Women's Health Initiative randomised trial. Diabetologia. 2006 doi: 10.1007/s00125-005-0096-0. Jan 27:1-10 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 65.Shionoiri H, Eggena P, Barrett JD. An increase in high molecular - weight rennin substrate associated with estrogenic hypertension. Biochem Med. 1983;29:14–22. doi: 10.1016/0006-2944(83)90049-2. [DOI] [PubMed] [Google Scholar]
- 66.Hodis HN, Mack WJ, Lobo RA, et al. Estrogen in the prevention of atherosclerosis. A randomized, double blind, placebo controlled trial. Ann Intern Med. 2001;135:939–953. doi: 10.7326/0003-4819-135-11-200112040-00005. [DOI] [PubMed] [Google Scholar]
- 67.Herrington DM, Reboussin DM, Brosniham KB, et al. Effects of estrogen replacement on the progression of coronary artery atherosclerosis. N. Engl J. Med. 2000;343:522–529. doi: 10.1056/NEJM200008243430801. [DOI] [PubMed] [Google Scholar]
- 68.Mueck AO, Seeger H, Wallweiner D. Medroxyprogesterone acetate versus norethisterone: effect on estradiol induced changes of markers for endothelial function and atherosclerotic plaque characteristics in human female coronary endothelial cell cultures. Menopause. 2002;9:273–281. doi: 10.1097/00042192-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 69.Pradham D, Mannon JE, Roussouw JE, et al. Inflammatory biomarkers, hormone replacement therapy and incident coronary heart disease. Prospective analysis from the Women's Health Initiative observational study. JAMA. 2002;28:980–987. doi: 10.1001/jama.288.8.980. [DOI] [PubMed] [Google Scholar]
- 70.Reuben DB, Palla SL, Hu P, et al. Progestins affect mechanism of estrogen-induced C-reactive protein stimulation. Am J Med. 2006;119:167.e1–167.e8. doi: 10.1016/j.amjmed.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 71.Lakoski SG, Herrington DM. Effects of hormone therapy on C-reactive protein and IL-6 in postmenopausal women: a review article. Climacteric. 2005;8:317–326. doi: 10.1080/13697130500345109. [DOI] [PubMed] [Google Scholar]
- 72.McKenzie J, Jaap AJ, Gallacher S, et al. Metabolic, inflammatory and haemostatic effects of a low-dose continuous combined HRT in women with type 2 diabetes: potentially safer with respect to vascular risk? Clin Endocrinol (Oxf) 2003;59:682–689. doi: 10.1046/j.1365-2265.2003.01906.x. [DOI] [PubMed] [Google Scholar]
- 73.Schlegel W, Petersdorf LI, Junker R, et al. The effects of six months of treatment with a low-dose of conjugated oestrogens in menopausal women. Clin Endocrinol (Oxf) 1999;51:643–651. doi: 10.1046/j.1365-2265.1999.00857.x. [DOI] [PubMed] [Google Scholar]
- 74.Notelovitz M. Hormone therapy and hemostasis. In: Lobo RA, editor. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. Second Edition. Philadelphia, Pa: Lippincott, Williams and Wilkens; 1999. p. 397. [Google Scholar]
- 75.Blann AD, Lip GY. Venous thromboembolism. BMJ. 2006;332:215–219. doi: 10.1136/bmj.332.7535.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith NL, Heckbert SR, Lemaire RN, et al. Esterified estrogen and conjugated equine estrogens in the risk of venous thrombosis. JAMA. 2004;292:1581–1587. doi: 10.1001/jama.292.13.1581. [DOI] [PubMed] [Google Scholar]
- 77.Scarabin P-Y, Oger E, Plu-Bureau G. Differential association of oral and transdermal oestrogen-replacement therapy with thromboembolism risk. Lancet. 2003;362:428–432. doi: 10.1016/S0140-6736(03)14066-4. [DOI] [PubMed] [Google Scholar]
- 78.Cano A, Van Baal WM. The mechanism of thrombotic risk induced by hormone replacement therapy. Maturitas. 2001;40:17–38. doi: 10.1016/s0378-5122(01)00270-5. [DOI] [PubMed] [Google Scholar]
- 79.Cushman M, Kuller LH, Prentice P, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292:1573–1580. doi: 10.1001/jama.292.13.1573. [DOI] [PubMed] [Google Scholar]
- 80.Dias AR, Jr, Melo RN, Gebara OC, et al. Effects of conjugated equine estrogens or raloxifene on lipid profile, coagulation and fibrinolysis factors in postmenopausal women. Climacteric. 2005;8:63–70. doi: 10.1080/13697130500042581. [DOI] [PubMed] [Google Scholar]
- 81.Cosman F, Baz-Hecht M, Cushman M, et al. Short-term effects of estrogen, tamoxifen and raloxifene on hemostasis: a randomized-controlled study and review of the literature. Thromb Res. 2005;116:1–13. doi: 10.1016/j.thromres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Pripp U, Eriksson-Berg M, Orth-Gomer K, et al. Does body mass index, smoking, lipoprotein levels, surgically induced menopause, hormone replacement therapy, years since menopause, or age affect hemostasis in postmenopausal women? Gend Med. 2005;2:88–95. doi: 10.1016/s1550-8579(05)80015-4. [DOI] [PubMed] [Google Scholar]
- 83.Matzdorff A. Platelet function tests and flow cytometry to monitor antiplatelet therapy. Semin Thromb Hemost. 2005;31:393–399. doi: 10.1055/s-2005-916672. [DOI] [PubMed] [Google Scholar]
- 84.Collaborative group on hormone factors in breast cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350:1047–1059. [PubMed] [Google Scholar]
- 85.Clemmons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2002;344:276–285. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 86.Lawson JS, Field AJ, Champion S, et al. Low estrogen receptor é expression in normal breast tissue underlies low breast cancer incidence in Japan. Lancet. 1999;354:1787–1788. doi: 10.1016/s0140-6736(99)04936-3. [DOI] [PubMed] [Google Scholar]
- 87.Chetrite GS, Pasqualini JR. The selective estrogen enzyme modulator (SEEM) in breast cancer. J Steroid Biochem Molec Biol. 2001;76:95–104. doi: 10.1016/s0960-0760(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 88.Clarke RB. Human breast proliferation and its relationship to steroid receptor expression. Climacteric. 2004;7:129–137. doi: 10.1080/13697130410001713751. [DOI] [PubMed] [Google Scholar]
- 89.Allred DC, Mohsin SK. Biological features of premalignant disease in the human breast. J Mammary Gland Biol Neoplasia. 2000;5:351–364. doi: 10.1023/a:1009573710675. [DOI] [PubMed] [Google Scholar]
- 90.Russo IH, Russo J. Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia. 1998;3:49–61. doi: 10.1023/a:1018770218022. [DOI] [PubMed] [Google Scholar]
- 91.Warren R. Hormones and mammographic breast density. Maturitas. 2004;49:67–78. doi: 10.1016/j.maturitas.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 92.Rutter CM, Mandelson MT, Laya MB, et al. Changes in breast density associated with initiation, discontinuation and continuing use of hormone replacement therapy. JAMA. 2001;285:171–176. doi: 10.1001/jama.285.2.171. [DOI] [PubMed] [Google Scholar]
- 93.Speroff L. The meaning of mammographic breast density in users of postmenopausal hormone therapy. Maturitas. 2002;41:171–175. doi: 10.1016/s0378-5122(02)00006-3. [DOI] [PubMed] [Google Scholar]
- 94.Junkermann H, van Holst T, Lang E, Rakov V. Influence of different HRT regimens on mammographic density. Maturitas. 2005;50:105–110. doi: 10.1016/j.maturitas.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 95.Yaffe M, Hendrix S, Pike M, et al. Is mammographic density, as currently measured, a robust surrogate marker for breast cancer? Gynecol Endocrinol. 2005;21(Suppl 1):17–21. doi: 10.1080/09513590400030004. [DOI] [PubMed] [Google Scholar]
- 96.O'Brien SN, Anandjiwala J, Price TM. Differences in estrogen content of breast adipose tissue in women by menopausal status and hormone use. Obstet Gynecol. 1997;90:244–248. doi: 10.1016/S0029-7844(97)00212-3. [DOI] [PubMed] [Google Scholar]
- 97.Cummings SR, Duong T, Kenyon E, et al. Serum estradiol level and risk of breast cancer during treatment with raloxifene. JAMA. 2002;287:216–220. doi: 10.1001/jama.287.2.216. [DOI] [PubMed] [Google Scholar]
- 98.Writing group for the Women's Health Initiative investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 99.Million Women study collaborators. Breast cancer and hormone replacement therapy in the Million Women study. Lancet. 2003;362:419–427. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 100.Shozu M, Sebastian S, Takayama K, et al. Estrogen excess associated with novel gain-of-function mutations affecting the aromatase gene. N Engl J Med. 2003;348:1855–1865. doi: 10.1056/NEJMoa021559. [DOI] [PubMed] [Google Scholar]
- 101.Hofseth LJ, Raafat AM, Osuch JR, et al. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84:4559–4565. doi: 10.1210/jcem.84.12.6194. [DOI] [PubMed] [Google Scholar]
- 102.Suparto IH, Williams JK, Cline JM, et al. Contrasting effects of the hormone replacement therapies on the cardiovascular and mammary gland outcomes in surgically menopausal monkeys. Am J Obstet Gynecol. 2003;188:1132–1140. doi: 10.1067/mob.2003.237. [DOI] [PubMed] [Google Scholar]
- 103.Isaksson E, Wang H, Sahlin B, et al. Effects of long-term HRT and tamoxifen on the expression of progesterone receptors A and B in breast tissue from surgically cynomolgus macaques. Breast Cancer Res Treat. 2003;79:233–239. doi: 10.1023/a:1023925906199. [DOI] [PubMed] [Google Scholar]
- 104.Silverman SL, Delmas PD, Kulkarni PM, et al. Comparison of fracture, cardiovascular event and breast cancer rates at 3 years in postmenopausal women with osteoporosis. J Am Geriatr Soc. 2004;52:154. doi: 10.1111/j.1532-5415.2004.52420.x. [DOI] [PubMed] [Google Scholar]