

Abstract
Atherosclerotic lesions form at distinct sites in the arterial tree, suggesting that hemodynamic forces influence the initiation of atherogenesis. If NF-κB plays a role in atherogenesis, then the activation of this signal transduction pathway in arterial endothelium should show topographic variation. The expression of NF-κB/IκB components and NF-κB activation was evaluated by specific antibody staining, en face confocal microscopy, and image analysis of endothelium in regions of mouse proximal aorta with high and low probability (HP and LP) for atherosclerotic lesion development. In control C57BL/6 mice, expression levels of p65, IκBα, and IκBβ were 5- to 18-fold higher in the HP region, yet NF-κB was activated in a minority of endothelial cells. This suggested that NF-κB signal transduction was primed for activation in HP regions on encountering an activation stimulus. Lipopolysaccharide treatment or feeding low-density lipoprotein receptor knockout mice an atherogenic diet resulted in NF-κB activation and up-regulated expression of NF-κB-inducible genes predominantly in HP region endothelium. Preferential regional activation of endothelial NF-κB by systemic stimuli, including hypercholesterolemia, may contribute to the localization of atherosclerotic lesions at sites with high steady-state expression levels of NF-κB/IκB components.
Keywords: atherosclerosis, p65, IκBα, VCAM-1, E-selectin
Atherosclerosis is a complex and chronic disease process involving elastic and muscular arteries. It is potentiated by systemic factors, including lipoprotein disorders, hypertension, diabetes mellitus, and smoking (1). Although clinically significant complications of atherosclerosis, such as plaque ulceration, rupture, and thrombosis, occur in established or advanced atherosclerotic plaques, elucidation of pathogenic mechanisms of early lesion formation may lead to interventions that delay or prevent lesion progression and complications. During initiation and expansion of fatty streaks, circulating monocytes are recruited to the arterial intima (2, 3). The arterial endothelium in these regions is activated and expresses inducible leukocyte adhesion molecules and chemokines.
Atherosclerotic lesions form at distinct sites in the arterial tree, especially at or near branch points, bifurcations, and curvatures. This distribution pattern suggests that local factors, such as hemodynamic forces (shear stress) that can be sensed by endothelial cells lining the artery wall, influence the initiation of atherogenesis. Blood flow is disturbed at branch points, bifurcations, and curvatures, unlike straight segments where it tends to be laminar (4). Alterations in shear stress can alter endothelial cell gene expression. For example, introduction of shear stress activates various signal transduction pathways in cultured endothelium (5, 6) and modulates the expression of adhesion molecules (7, 8). Different shear stress profiles can induce distinct repertoires of endothelial gene expression (9). Hemodynamic forces may also influence topographic variations in gene expression by endothelial cells in arteries and may predispose regions to atherosclerotic lesion formation if appropriate systemic risk factors are present.
The NF-κB signal transduction pathway mediates increased expression of many cytokine-induced genes that protect cells from apoptosis and participate in inflammatory and immune processes (10, 11). This pathway may also participate in atherogenesis. Previous studies demonstrated NF-κB activation in established human atherosclerotic plaques (12), and many genes expressed in plaques are regulated by NF-κB (13). In normocholesterolemic animals, we reported higher expression of vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule (ICAM)-1 in endothelial cells in regions of the mouse ascending aorta and arch predisposed to lesion formation (14). Cytokine-induced expression of these adhesion molecules is regulated by NF-κB (15) and suggests that NF-κB activation may participate in the initiation of lesions. Hemodynamics may be involved, because NF-κB is activated in cultured endothelium by acute alterations of shear stress (6, 16–19) and in regions of disturbed flow (20).
The NF-κB/Rel family of proteins consists of homo- or heterodimers [reviewed by Ghosh and Karin (10, 11)]. Subunits include NF-κB1 (p50), NF-κB2 (p52, p49, p50B), p65 (RelA), RelB, and c-Rel. All are expressed ubiquitously except for RelB and c-Rel, which are largely restricted to lymphoid and hematopoietic cells, respectively. In cultured endothelial cells, p50/p65 is the predominant NF-κB species (21). In quiescent cells, NF-κB is localized in the cytoplasm, where it is retained through association with an inhibitor (10, 11). Inhibitors of NF-κB (IκBs) include IκBα, IκBβ, IκBɛ, bcl-3, p105 (precursor of p50) and p100 (precursor of p52) and IκBγ (a 70-kDa protein arising by alternate promoter usage in the p105 gene). IκBs contain six or more ankyrin repeats, a N-terminal regulatory domain, and a C-terminal domain that contains a PEST motif involved in basal turnover. Different IκBs bind preferentially to different NF-κB dimers and sterically hinder binding of importins to the nuclear localization sequence of NF-κB subunits.
Diverse stimuli can activate NF-κB through phosphorylation and activation of IκB kinase complex (10, 11). Activated IκB kinases phosphorylate IκBs leading to their polyubiquitination and degradation by 26S proteasomes. NF-κB dimers are transported to the nucleus, where they bind to the major groove of DNA and transactivate gene expression through interactions with other transcription factors and coactivators (22). Each NF-κB subunit has different DNA-binding and transactivation properties. Activation of NF-κB provides negative feedback to this signaling pathway by rapidly up-regulating IκBα expression, resulting in replenishment of the cytoplasmic pool of its inhibitor. IκBα contains a nuclear export sequence and after associating with DNA-bound NF-κB, it binds exportins and transports NF-κB back to the cytoplasm (10, 11).
The extent of atherosclerotic lesion formation in mice deficient in either apolipoprotein E or LDL receptor (LDLR−/−) is quite variable, yet the location of lesions particularly in the ascending aorta is highly reproducible (23, 24). In a previous study we used en face oil red O staining to map regions of the LDLR−/− mouse ascending aorta and arch that were highly predisposed or protected from atherosclerotic lesion formation (14), and designated these as high and low probability (HP and LP) regions. The goal of the current study was to investigate the possibility that NF-κB signal transduction in endothelium contributes to the localization of atherosclerotic lesions. Immunoconfocal microscopy of aortic endothelium of normocholesterolemic mice demonstrated that the expression of p65 and key IκBs was markedly and selectively elevated in HP regions, yet NF-κB activation was low, suggesting that the NF-κB pathway was primed for activation. Systemic treatment with graded doses of lipopolysaccharide (LPS) or feeding LDLR−/− mice a cholesterol-enriched diet for 2 weeks resulted in preferential NF-κB activation and induction of NF-κB responsive gene expression in HP, but not LP, regions.
Materials and Methods
Animals and Treatments.
C57BL/6 and LDLR−/− mice backcrossed 10 generations into the C57BL/6 strain were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained in accordance with guidelines of the Canadian Council on Animal Care. Experiments were carried out at 2 to 4 months of age using (i) untreated standard chow-fed wild-type and LDLR−/− mice, (ii) endotoxin-treated mice (0, 10 or 100 μg i.p. Escherichia coli 055:B5 LPS, Sigma) and (iii) LDLR−/− mice fed for 2 weeks an AIN-76A semipurified cholate-free diet containing high fat (40% of energy intake) and 1.25% cholesterol (Research Diets, New Brunswick, NJ, diet no. D12108) (25). Aortas were harvested after perfusion with PBS and 2% paraformaldehyde (14). For silver nitrate staining, aortas were perfused with PBS, 0.25% silver nitrate, PBS again, and 2% paraformaldehyde via the left ventricle (see supplementary material, www.pnas.org).
Immunofluorescence Staining Reagents and Protocols.
Primary antibodies included anti-p65 (sc-372-G), anti-IκBα (sc-371-G), anti-IκBβ (sc-946), and anti-PECAM-1 (sc-1506) from Santa Cruz, phospho-IκBα (Ser32) from New England Biolabs, anti-mouse E-selectin and anti-mouse ICAM-2 from PharMingen, and anti-mouse VCAM-1 (M/K-2.7) from American Type Culture Collection. Negative controls by using the distal arch included nonimmune goat, rabbit, or rat IgG (Jackson ImmunoResearch Labs). For anti-p65, the primary antibody was incubated with blocking peptide-immunogen (sc-372P, Santa Cruz, 1:50 dilution). Secondary antibodies included biotin-conjugated donkey polyclonal anti-goat, -rabbit, or -rat IgG (Jackson), or Cy3-labeled donkey anti-goat or anti-rat.
Tyramide signal amplification (NEN) was used for detection of p65, IκBα, IκBβ, phospho-IκBα, E-selectin, and ICAM-2 according to the manufacturer's protocol, and Cy3-labeled secondary antibodies were used to detect VCAM-1 and PECAM-1 expression (14). Immunostaining included blocking of endogenous peroxidase with 3% H2O2, permeabilization with 0.2% Triton X-100, and blocking with Tris⋅HCl blocking buffer. Primary antibody incubations were for 1 h at 22°C for p65, IκBα, IκBβ and ICAM-2, and overnight at 4°C for E-selectin and phospho-IκBα. Subsequent incubations were biotin-conjugated secondary antibody (30 min), steptavidin-conjugated horseradish peroxidase (30 min) and FITC- or Cy3-conjugated tyramide complexes (8 min). Tris⋅HCl blocking buffer (NEN) was used for diluting antibodies and for washes. Nuclei were counterstained with Propidium Iodide (Molecular Probes) or Sytox (Molecular Probes).
Confocal Microscopy.
The ascending aorta and arch were opened and mounted with the endothelium facing up (14). Images of the endothelial cell monolayer were obtained by using a Bio-Rad MRC-1024ES confocal microscope equipped with a krypton/argon laser and a 60 × 1.4-numerical aperture objective (Nikon). HP and LP regions were located by using three anatomic landmarks as reference points (14) (supplementary Fig. 5; see www.pnas.org). In every mouse, three or four images were obtained from each HP and LP region. An HP region was sampled first and was used to optimize the confocal settings, which were maintained for other images, including the negative control (distal arch). The distribution and intensity of fluorescence were quantified for each antibody by using the confocal software frequency histogram function. For each mouse, the negative control was used to establish a pixel intensity that eliminated 99% of the background signal. Background fluorescence was then subtracted by applying this threshold to all HP and LP images, and the percent pixels with remaining signal and the average signal intensity were recorded for each image.
Statistical Analyses.
Differences among treatment groups were evaluated by using a one-way ANOVA. Within each treatment group, a paired t test was used to determine differences between HP and LP regions.
Results and Discussion
Endothelial Cells in the HP Region Have a Polygonal Shape and a Random Orientation.
In our experience with LDLR−/− mice, the extent of atherosclerotic lesion formation is variable even among inbred littermates; however, the location of lesions is highly reproducible, particularly in the ascending aorta and arch. Other investigators have reported similar observations in mice and other species and have attributed this to regional differences in hemodynamics. The shape of endothelial cells reflects the local hemodynamic environment (26); therefore, we stained intercellular junctions with silver nitrate to visualize endothelium in the HP and LP regions of the mouse proximal aorta, which we mapped previously (14). Endothelial cells in the LP region were elongated parallel to the direction of blood flow, whereas in the HP region they were more polygonal and oriented randomly (Fig. 1). Endothelial nuclei in the LP region were oval, parallel to the direction of blood flow, and uniformly spaced. In the HP region, they were irregular and appeared to be at a higher density (Figs. 2–4). These observations are indicative of disturbed and laminar hemodynamic flow patterns in HP and LP regions, respectively, based on studies demonstrating similar endothelial morphology after exposure to disturbed or laminar flow (4, 24). Endothelial cells can also adopt a polygonal shape when shear stresses are very low, but this is unlikely to be the case in the aortic arch.
Figure 1.

Silver nitrate staining in HP and LP regions. Representative images were obtained from a C57BL/6 mouse aorta. Endothelial cells in the HP region have variable shapes and random orientation, whereas in the LP region they are elongated and aligned in the direction of blood flow (Left to Right).
Figure 2.

High expression levels of p65, IκBα, and IκBβ in the HP region of untreated standard chow-fed C57BL/6 mice. (a) For each antibody, representative confocal images of HP and LP regions obtained from the same mouse are shown (p65 and IκBα, green and IκBβ, red fluorescence). (b) Fluorescent staining is quantified for each antibody in histograms that illustrate the percent of pixels with a positive signal after subtraction of background fluorescence. Similar expression levels of p65 were found in standard chow-fed LDLR−/− mice bred into the C57BL/6 background (LDLR−/−) and wild-type C57BL/6 mice (LDLR+/+). Data are presented as means ± SD. Significant differences (P < 0.05) were found between all HP and LP regions, determined by using a paired t test (n = 5).
Figure 4.

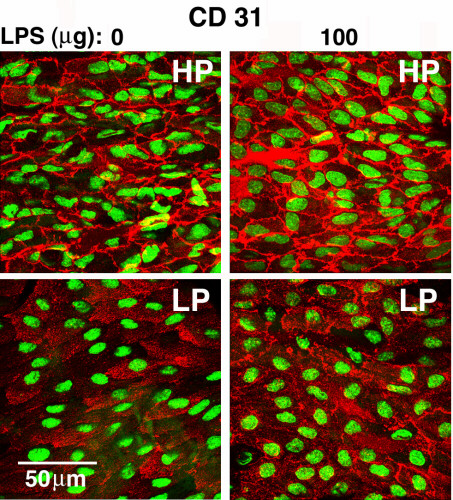
Constitutive and LPS-induced expression of VCAM-1, E-selectin, and ICAM-2 in HP and LP regions. (a) Representative immunoconfocal images of permeabilized endothelium illustrate LPS-induced VCAM-1 (red) and E-selectin (green) but not ICAM-2 (green) expression. The magnitude of LPS-induced expression is significantly greater in HP regions. In control mice (0 μg LPS), VCAM-1, but not E-selectin, expression is detected in the HP region. Expression of ICAM-2 and PECAM-1 (CD 31) (supplementary Fig. 7; see www.pnas.org) is abundant in HP and LP regions of control and LPS-treated mice and is not induced by LPS. (b) The means ± SD of percent pixels with fluorescent staining are plotted for each group (n = 3–5). There are significant differences (P < 0.05, paired t test) between HP and LP regions for VCAM-1 and E-selectin but not for ICAM-2 and PECAM-1. Significant differences from 0 and 10 μg LPS groups are denoted by * and †, respectively (ANOVA).
Expression Levels of p65, IκBα, and IκBβ Are High in the HP Region of Untreated Standard Chow-Fed Mice, Yet NF-κB Activation Is Relatively Low.
In endothelial cells, the main form of NF-κB with transactivation potential is p65, and in resting cells it is associated predominantly with IκBα and IκBβ (21). We used a polyclonal antibody that recognizes p65 in the cytoplasm of resting cells when complexed to IκBs as well as when translocated to the nucleus on activation of NF-κB. Initial experiments demonstrated the specificity of immunostaining of mouse aortic endothelium with this anti-p65 antibody and showed that inclusion of the blocking peptide (immunogen) during incubations with primary antibody completely inhibited staining (supplementary Fig. 6; see www.pnas.org). In the ascending aorta and proximal arch, the HP and LP regions were located in the lesser and greater curvatures, respectively (14), which permitted meaningful comparison of fluorescent signal intensities after immunostaining of the entire aortic segment (supplementary Fig. 5).
Endothelial cell expression of p65, IκBα, and IκBβ was 5- to 18-fold greater in the HP vs. the LP region of untreated C57BL/6 and standard chow-fed LDLR−/− mice (Fig. 2). The extent of NF-κB activation was determined by nuclear translocation of p65 and staining for serine32 phosphorylated IκBα (phospho-IκBα). Nuclear translocation of p65 was found in 12.5 ± 5.5% endothelial cells of the HP region vs. 2.4 ± 4.4% in the LP region (P < 0.0001 paired t-test, n = 10). Translocation was comparable in wild-type C57BL/6 and LDLR−/− mice fed standard lab chow. Phospho-IκBα staining was detected as occasional discrete cytoplasmic aggregates and was more abundant in the HP vs. LP region (4.7 ± 1.2 vs. 0.43 ± 0.28% positive pixels, P < 0.05, n = 3), consistent with the p65 nuclear translocation data. Although NF-κB activation was significantly greater in the HP vs. the LP region, only a fraction of the cytoplasmic p65 was translocated, and this occurred in <15% of HP region endothelial cells.
The differences in endothelial cell expression levels of NF-κB/IκB components in the HP and LP regions may be the result of unique hemodynamic properties in these regions, which would be consistent with different endothelial cell morphology. However, a direct role for hemodynamics in NF-κB/IκB expression has not been established by our study and has not been evaluated in cell-culture models. Therefore, other possibilities should not be overlooked such as early events associated with atherogenesis. In normocholesterolemic animals, increased retention of LDL has been found in lesion-prone vs. lesion-resistant regions of the aorta (27, 28), and in human fetal aortas, there is increased intimal accumulation of oxidized LDL in lesion-prone sites (29).
Although NF-κB can be activated in cultured endothelium by acute alterations of shear stress (6, 16–19) and by flow disturbances (20), these experiments did not address how prolonged alterations in shear stress or flow profiles affect activation. We found that NF-κB was activated in a minority of HP region endothelial cells, which suggests that regional hemodynamic factors do not provide critical activation signals, although they may be responsible for the different expression levels of NF-κB/IκB components.
LPS Treatment or Feeding LDLR−/− Mice an Atherogenic Diet Activates NF-κB Predominantly in the HP Region.
The above data demonstrated that in untreated standard chow-fed mice, NF-κB was activated in only a minority of endothelial cells located predominantly in the HP region. Most HP region endothelial cells contained abundant cytoplasmic p65 and IκBs, and this suggested that their NF-κB signal transduction pathway was primed for activation. If these cells encounter a stimulus that activates IκB kinases, abundant IκB substrate would be available for phosphorylation and degradation, allowing multiple molecules of p65 to translocate to the nucleus. Expression of NF-κB components was relatively low in the LP region; therefore, the extent of p65 nuclear translocation and gene transactivation would be low even after a potent activation stimulus. This possibility was evaluated by injecting wild-type C57BL/6 mice with LPS or by feeding LDLR−/− mice with an atherogenic diet for 2 weeks.
NF-κB activation has been reported in advanced human atherosclerotic lesions (12), but activation in endothelium of early lesions and before lesion formation has not been evaluated. Feeding LDLR−/− mice a cholesterol-enriched diet models an important systemic risk factor and reproducibly produces atherosclerotic lesions. Based on studies that demonstrated increased lipoprotein retention at atherosclerosis-prone sites (27), it is possible that an atherogenic diet could provide preferential activation signals in the HP region. LPS, an agent known to activate NF-κB in a variety of cells (10, 11), was used to provide an acute systemic activation stimulus in both HP and LP regions. Although the contribution to atherogenesis of such an acute systemic activation of endothelium is not known, inflammatory mediators produced by acute or chronic infections are thought to modulate the development and complications of atherosclerotic lesions.
LPS treatment resulted in 6- to 10-fold increased cytoplasmic phospho-IκBα staining in both HP and LP regions (Fig. 3). This suggests that IκB kinases were activated comparably. The lower absolute levels of phospho-IκBα in the LP region can be explained by the lower content of IκBα in these cells. The extent of phospho-IκBα staining in the LP region of LPS-treated mice was comparable to that found in the HP region of controls (Fig. 3b). LPS induced a significant and dose-dependent nuclear translocation of p65 in endothelium of the HP region (Fig. 3). Staining for p65 covered virtually the entire nucleus, and cytoplasmic staining was reduced or absent. In contrast, endothelial nuclei in the LP region did not show a significant increase in p65 translocation and showed only focal positive staining. The lack of a small increase in p65 translocation in the LP region induced by LPS may reflect control of nuclear translocation in the LP region. Alternatively, it may have been an experimental artifact, because the accuracy of quantifying low-level p65 fluorescent signals is diminished as a result of background autofluorescence from the internal elastic lamina. This may explain the high variability in the control group (0 μg LPS). Overall, the LPS studies showed that the relatively high levels of NF-κB/IκB components in the HP region were functionally quite responsive, whereas the NF-κB signal transduction pathway in the LP region was largely quiescent.
Figure 3.

LPS treatment or feeding LDLR−/− mice an atherogenic diet activates NF-κB predominantly in the HP region. (a) Representative immunoconfocal images from HP and LP regions are shown. Punctate green staining for phospho-IκBα (P-IκBα) is abundant in the cytoplasm of HP region endothelium after administration of 100 μg LPS. Nuclear translocation of p65 (green) is seen in HP regions 30 min after i.p. injection of LPS. Nuclei are counterstained with propidium iodide. Increased p65 nuclear translocation occurs in HP regions of LDLR−/− mice 2 weeks after ingestion of a 1.25% cholesterol-enriched diet (CHOL.) relative to standard chow-fed (CHOW) mice. (b) Fluorescent staining is quantified in histograms showing percent of pixels positive for phospho-IκBα and percent of nuclei with translocated p65 (mean ± SD, n = 3 to 5). Significant differences (P < 0.05) are found between all HP and LP regions (paired t test). Significant differences from 0 and 10 μg LPS and chow groups are denoted by *, †, and ‡, respectively (ANOVA).
Feeding LDLR−/− mice a cholesterol-enriched diet for 2 weeks resulted in a 2-fold increase in p65 nuclear translocation in the HP region as compared with mice receiving standard lab chow (Fig. 3). At this time point, oil red O-stainable lesions had not developed in the HP region, but it is possible that occasional monocytes began accumulating in the intima. In the LP region, a significant increase in p65 nuclear translocation was also observed. These data suggest that hypercholesterolemia provides an activation stimulus that is not restricted to regions predisposed to atherosclerotic lesion formation. This is consistent with recent data from rats injected with a single bolus of human LDL demonstrating its accumulation and oxidation throughout the aorta, associated with NF-κB activation and up-regulation of ICAM-1 expression in endothelium (30). In the LP region of cholesterol-fed LDLR−/− mice, the extent of endothelial cell p65 nuclear translocation was 4- to 5-fold lower than that found in the HP region of standard chow-fed LDLR−/− mice (2.7% vs. 12.8% of nuclei). This suggests that the magnitude of NF-κB activation in aortic endothelium depends on the expression levels of NF-κB/IκB components. It is interesting to note that cholesterol feeding did not increase the expression levels of p65 or IκBs.
Expression of NF-κB Target Genes Is Preferentially Up-Regulated by LPS in the HP Region.
The functional consequences of NF-κB activation were assessed by evaluating the expression of VCAM-1 and E-selectin, genes whose induced expression depends on NF-κB activation (15). Aortas from C57BL/6 mice were harvested 5 or 6 h after LPS injection for E-selectin and VCAM-1, respectively, and immunostaining was carried out on Triton X-100-permeabilized endothelium. LPS treatment induced a dose-dependent increase in expression of both VCAM-1 and E-selectin in the HP region, whereas in the LP region, low expression levels were found even at the 100 μg dose (Fig. 4). For example, the expression level of VCAM-1 induced by the 100 μg dose of LPS in the LP region did not approach that found in the HP region of control mice (Fig. 4b). These results suggest a strong correlation between nuclear translocation of p65 and expression of VCAM-1 and E-selectin. Differences between VCAM-1 and E-selectin expression (VCAM-1 was expressed preferentially in the HP region of control mice, whereas E-selectin expression was not detected) may reflect different sensitivities of these genes to NF-κB activation.
ICAM-2 and PECAM-1 were selected as negative controls for the above experiments, because the expression of these genes by endothelial cells is constitutive, not up-regulated by LPS or cytokines (31, 32), and thought to be independent of NF-κB activation. The expression levels of PECAM-1 or ICAM-2 in HP and LP regions were comparable in untreated mice and were not altered 12 h after a 100 μg LPS injection (Fig. 4). ICAM-2 localization was similar in endothelial cells of HP and LP regions, unlike PECAM-1. In the HP region, PECAM-1 was concentrated at intercellular junctions, whereas it was also distributed throughout the cytoplasm in the LP region. This pattern was not affected by LPS treatment (supplementary Fig. 7; see www.pnas.org).
Our studies indicated that the NF-κB signal transduction pathway in endothelium of HP regions was primed to respond to systemic activation stimuli, including ingestion of an atherogenic diet. The HP and LP regions that we studied likely represent two extremes of a spectrum. Our preliminary observations of the mouse descending thoracic aorta support this concept in that p65 staining in a random segment of the descending thoracic aorta was intermediate to that of HP and LP regions (supplemental Fig. 6). It would be interesting to speculate whether the NF-κB signal transduction pathway is up-regulated in HP regions or down-regulated in LP regions. Acute exposure of endothelium to laminar shear stress can activate various signal-transduction cascades and lead to alterations in expression levels of numerous genes (33). After endothelial cells become acclimatized to an alteration in laminar shear forces, these signaling cascades are down-regulated; however, it is possible that some signal-transduction pathways are chronically active in HP regions which have complex or disturbed hemodynamics. In vivo most vascular endothelial cells, including those in arterial regions with low predisposition to atherosclerosis, are chronically exposed to laminar shear forces, which is their physiological milieu. Chronic exposure to laminar shear stress may up-regulate the expression of unique genes that are atheroprotective (34, 35), or may promote mechanisms that actively repress expression of broad categories of genes. Several groups have demonstrated that surgically introduced alterations in fluid dynamics in conjunction with hypercholesterolemia influence atherosclerotic lesion formation (36, 37), which further supports the role of hemodynamics in lesion development.
In parallel to NF-κB, it is likely that other signal transduction pathways or transcription factors are up-regulated by vascular cells in a regional fashion. For example, Egr-1 is a transcription factor that can be induced by acute stimuli, including shear stress, and is up-regulated in the intima of atherosclerotic lesions (38). Regional differences in only a few signaling pathways or transcription factors may result in dramatically different biological responses to systemic activation stimuli, because each pathway and transcription factor may regulate the expression of multiple genes.
Supplementary Material
Acknowledgments
We thank Dr. Lowell Langille for his advice and comments on the manuscript. This work was supported by the Heart and Stroke Foundation of Ontario (grant T-3588), the Medical Research Council of Canada (grant MT-14151), and the National Institutes of Health (grant P50 HL56985). M.C. holds an Established Investigatorship from the American Heart Association.
Abbreviations
- HP
high probability
- LP
low probability
- LDLR
low density lipoprotein receptor
- LPS
lipopolysaccharide
- VCAM-1
vascular cell adhesion molecule 1
- ICAM
intercellular adhesion molecule
- PECAM-1
platelet endothelial cell adhesion molecule 1 (CD31)
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Fuster V, Ross R, Topol E J. Atherosclerosis and Coronary Artery Disease. Philadelphia: Lippincott–Raven; 1996. [Google Scholar]
- 2.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Munro J M, Cotran R S. Lab Invest. 1988;58:249–261. [PubMed] [Google Scholar]
- 4.Ku D N, Zhu C. In: Hemodynamic Forces and Vascular Cell Biology. Sumpio B E, editor. Austin, TX: Landes; 1993. pp. 1–23. [Google Scholar]
- 5.Ishida T, Takahashi M, Corson M A, Berk B C. Ann NY Acad Sci. 1997;811:12–23. doi: 10.1111/j.1749-6632.1997.tb51984.x. [DOI] [PubMed] [Google Scholar]
- 6.Bhullar I S, Li Y-S, Miao H, Zandi E, Kim M, Shyy J Y-J, Chien S. J Biol Chem. 1998;273:30544–30549. doi: 10.1074/jbc.273.46.30544. [DOI] [PubMed] [Google Scholar]
- 7.Nagel T, Resnick N, Atkinson W J, Dewey C F, Jr, Gimbrone M A., Jr J Clin Invest. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ando J, Tsuboi H, Korenaga R, Takada Y, Toyama-Sorimachi N, Miyasaka M, Kamiya A. Am J Physiol. 1994;267:C679–C687. doi: 10.1152/ajpcell.1994.267.3.C679. [DOI] [PubMed] [Google Scholar]
- 9.Gimbrone M A, Jr, Nagel T, Topper J N. J Clin Invest. 1997;100:S61–S65. [PubMed] [Google Scholar]
- 10.Ghosh S, May M J, Kopp E B. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 11.Karin M. J Biol Chem. 1999;274:27339–27342. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 12.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle P A, Neumeier D. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins T. Lab Invest. 1993;68:499–508. [PubMed] [Google Scholar]
- 14.Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff B D, Cybulsky M I. Circ Res. 1999;85:199–207. doi: 10.1161/01.res.85.2.199. [DOI] [PubMed] [Google Scholar]
- 15.Collins T, Read M A, Neish A S, Whitley M Z, Thanos D, Maniatis T. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 16.Khachigian L M, Resnick N, Gimbrone M A, Jr, Collins T. J Clin Invest. 1995;96:1169–1175. doi: 10.1172/JCI118106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao X, Lu C, Frangos J A. Arterioscler Thromb Vasc Biol. 1999;19:996–1003. doi: 10.1161/01.atv.19.4.996. [DOI] [PubMed] [Google Scholar]
- 18.Mohan S, Mohan N, Sprague E A. Am J Physiol. 1997;273:C572–C578. doi: 10.1152/ajpcell.1997.273.2.C572. [DOI] [PubMed] [Google Scholar]
- 19.Mohan S, Mohan N, Valente A J, Sprague E A. Am J Physiol. 1999;276:C1100–C1107. doi: 10.1152/ajpcell.1999.276.5.C1100. [DOI] [PubMed] [Google Scholar]
- 20.Nagel T, Resnick N, Dewey C F, Jr, Gimbrone M A., Jr Arterioscler Thromb Vasc Biol. 1999;19:1825–1834. doi: 10.1161/01.atv.19.8.1825. [DOI] [PubMed] [Google Scholar]
- 21.Read M A, Whitley M Z, Williams A J, Collins T. J Exp Med. 1994;179:503–512. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheppard K A, Rose D W, Haque Z K, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld M G, Glass C K, Collins T. Mol Cell Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakashima Y, Plump A S, Raines E W, Breslow J L, Ross R. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 24.Palinski W, Tangirala R K, Miller E, Young S G, Witztum J L. Arterioscler Thromb Vasc Biol. 1995;15:1569–1576. doi: 10.1161/01.atv.15.10.1569. [DOI] [PubMed] [Google Scholar]
- 25.Lichtman A H, Clinton S K, Iiyama K, Connelly P W, Libby P, Cybulsky M I. Arterioscler Thromb Vasc Biol. 1999;19:1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 26.Gimbrone M A., Jr Am J Pathol. 1999;155:1–5. doi: 10.1016/S0002-9440(10)65090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwenke D C. Arterioscler Thromb Vasc Biol. 1995;15:1928–1937. doi: 10.1161/01.atv.15.11.1928. [DOI] [PubMed] [Google Scholar]
- 28.Schwenke D C, Carew T E. Arteriosclerosis. 1989;9:908–918. doi: 10.1161/01.atv.9.6.908. [DOI] [PubMed] [Google Scholar]
- 29.Napoli C, D'Armiento F P, Mancini F P, Postiglione A, Witztum J L, Palumbo G, Palinski W. J Clin Invest. 1997;100:2680–2690. doi: 10.1172/JCI119813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calara F, Dimayuga P, Niemann A, Thyberg J, Diczfalusy U, Witztum J L, Palinski W, Shah P K, Cercek B, Nilsson J, et al. Arterioscler Thromb Vasc Biol. 1998;18:884–893. doi: 10.1161/01.atv.18.6.884. [DOI] [PubMed] [Google Scholar]
- 31.Henninger D D, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson D C, Granger D N. J Immunol. 1997;158:1825–1832. [PubMed] [Google Scholar]
- 32.Xu H, Bickford J K, Luther E, Carpenito C, Takei F, Springer T A. J Immunol. 1996;156:4909–4914. [PubMed] [Google Scholar]
- 33.Takahashi M, Ishida T, Traub O, Corson M A, Berk B C. J Vasc Res. 1997;34:212–219. doi: 10.1159/000159225. [DOI] [PubMed] [Google Scholar]
- 34.Topper J N, Cai J, Falb D, Gimbrone M A., Jr Proc Natl Acad Sci USA. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Traub O, Berk B C. Arterioscler Thromb Vasc Biol. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 36.Imparato A M, Lord J W, Jr, Texon M, Helpern M. Surg Forum. 1961;12:245–247. [Google Scholar]
- 37.Gyurko G, Szabo M. Surgery. 1969;66:871–874. [PubMed] [Google Scholar]
- 38.McCaffrey T A, Fu C, Du B, Eksinar S, Kent K C, Bush H, Jr, Kreiger K, Rosengart T, Cybulsky M I, Silverman E S, et al. J Clin Invest. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}