

Abstract
The endocrinology of the menopausal transition involves a complex interaction of molecular and tissue-specific hormone receptors, enzymes, and moderating cofactors that determine the functional expression of a given organ. The synthesis and metabolism of estrogen in estrogen-sensitive organs continue postmenopausally, albeit at levels substantially reduced from those of reproductive women. The postmenopausal production of estrogen is genetically determined. Thus, symptoms of estrogen deprivation will vary among menopausal women, although all will cease to menstruate. All prescribed estrogens have a similar class effect and exert their estrogenicity through similar genomic and nongenomic pathways. However, the source, chemical structure, and composition of the estrogens most commonly prescribed for menopausal complaints – conjugated equine estrogens (CEE), micronized 17beta estradiol (E2), and ethinyl estradiol (EE) – vary in content, pharmacokinetics, and pharmacodynamics. These variables are further influenced by dosage and route of administration. The net clinical effect depends on the type and amount of free bioavailable estrogen derived exogenously combined with the respective organ's endogenous synthesis of estrogen. Extrapolation of population- and group-based randomized clinical trials that evaluate a fixed dose of a standard estrogen preparation over a predetermined period of time may not be applicable to other products or to individual women whose biology differs from that of the study population. The decision to prescribe estrogen therapy for menopausal symptoms should be considered within the context of the woman's total quality of life healthcare needs and adjusted over time to ensure maximal efficacy with minimal risk.
Readers are encouraged to respond to George Lundberg, MD, Editor of MedGenMed, for the editor's eye only or for possible publication via email: glundberg@medscape.net
Introduction
Estrogen is a systemic steroid. In addition to its essential role in reproduction, estrogen acts physiologically to maintain the health and normal function of organs as diverse as the brain, heart, and bone. The amount of estrogen synthesized decreases significantly after menopause. Yet, although postmenopausal women are estrogen deficient relative to their premenopausal cohorts, they are not estrogen depleted. Postmenopausal estrogen synthesis and metabolism continue in target organs, with tissue levels of the steroids in excess of that measured peripherally.[1] The degree to which this occurs differs between individuals, and within an individual from organ to organ. In addition, each organ appears to have an undefined estrogen threshold, below which an estrogen-related symptom or function will manifest. This accounts in part, for the variable symptomatology noted in menopausal women[2] and differing response to a standard dose of estrogen therapy (ET).
The safety of postmenopausal ET has been questioned. Recommendations regarding the dosage, timing of initiation, and duration of exposure to ET have been revised, based in large measure by the outcome of a single randomized controlled trial (RCT) – The Women's Health Initiative (WHI).[3] The conclusions based on results of this study were considered equally applicable to all estrogens currently prescribed in clinical practice on the basis of an assumption that all estrogens and dosages are the same. However, the conclusions of the WHI are not entirely consistent with the large body of previously published RCT and observational studies of ET, results derived from basic scientific studies of ET, and, most importantly, with clinical experience.
The article reviews an approach to postmenopausal ET that is based on 3 main principles: (1) the pathogenesis of estrogen-associated conditions and diseases across a woman's life cycle; (2) the age-related endogenous synthesis of estrogen in postmenopausal women, and (3) the pharmacokinetics and pharmacodynamics of the 3 estrogens – CEE, E2, and EE – commonly prescribed for menopausal care. Simply stated, the clinical application of this biologic approach is to base the ET decision on (1) the presence (and persistence) of conditions related to estrogen deficiency; (2) titration of the dose of ET to meet clinical efficacy and safety (individualized ET); (3) adjustment of the dose of ET over time to account for the increased peripheral aromatization of androgen to estrogen with aging (age-adjusted ET); (4) timing the initiation of ET to optimize its efficacy and minimize adverse events; (5) annual monitoring of ET to ensure continued therapeutic efficacy and safety. The subject will be covered in 2 separate articles: the present article covers the biology of postmenopausal endogenous and exogenous estrogen activity and the management of the symptomatic menopause, and the companion article[4] covers applied sex steroid biology and long-term age-adjusted ET.
Estrogen Steroidogenesis and Action
The synthesis and biologic action of estrogen is complex.[1,5] E2 is the principal bioavailable ligand that binds to specific estrogen receptors (ERs), leading to the dimerization of the receptors, conformational change in the estrogen response elements, and the recruitment of co-regulator proteins. These co-activators and co-repressors influence the transcriptional activity of the activated ERs and the gene expression of the target cells. This cytoplasmic/nuclear interaction (classic pathway) takes minutes or hours. In addition to this process, estrogenic action can be activated more quickly via ERs bound to the cell surface or by estrogen-independent activation of the ERs.[1,5,6]
Synthesis and Metabolism of Estrogen
Postmenopausal estrogen is derived primarily from the bioconversion of androgens (from the adrenal gland and ovarian stroma) and, to a lesser extent, from the storage form of estrogen, estrone sulfate (E1S). The pathways of estrogen synthesis are summarized in the Figure.[1,5,7] Each step is catalyzed via a specific enzyme, which in turn is controlled by a specific gene. Aromatization of androgens to estrogen takes place primarily in the extragonadal tissues – liver, muscle, and fat – and increases with aging.[8] E2 synthesis occurs principally through oxidation of estrone (E1) and is a reversible reaction. The E2/E1 ratio before menopause is greater than 1; after menopause E1 is the predominant estrogen. E2 is transported in serum, reversibly bound to sex hormone-binding globulin (SHBG) and, with less affinity, to albumin. Both E2 and E1 can be metabolized to the biologically weaker estrogen, estriol, and to a number of other metabolites via A-ring metabolic pathways (eg, 2-hydroxyestrone and its methylated form 2-methoxyestrone) or D-ring metabolism (eg,16-alpha hydroxyestrone). These metabolic pathways are also genetically controlled (eg, COMT). The significance of these metabolites is their linkage to either anticarcinogenic (A-ring) or potential carcinogenic (D-ring) activity. Included in the latter category are the 4-hydroxyestrogens.[9]
Figure 1.
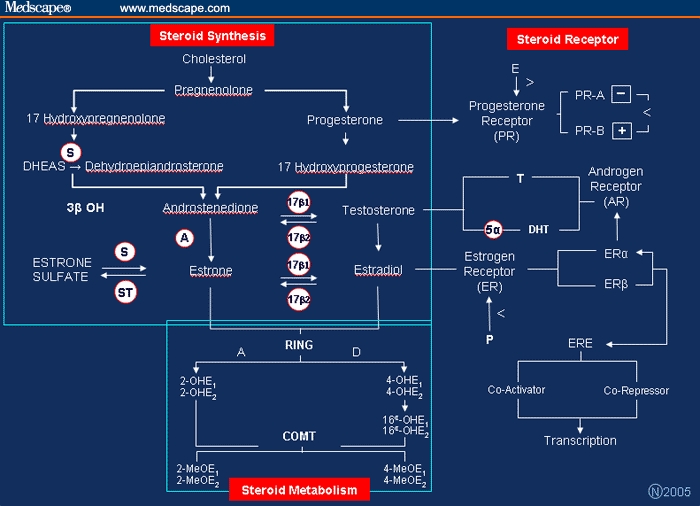
Pathways and enzymes in estrogen, progesterone, and testosterone biosynthesis, metabolism and steroid receptor activity.
A = aromatase activity
S = sulfatase activity
ST = sulfotransferase activity
17beta1 and 17beta2 = 17beta dehydrogenase activity type 1 and 2
3beta OH = 3beta hydroxysteroid dehydrogenase
> = receptor upregulation
< = receptor downregulation
OHE1 =2hydroxyestrone
OHE2 = 2hydroxyestradiol
MEOE1 = 2methoxyestrone
MEOE2 = and 2methoxyestradiol
5α = 5α reductase types 1 and 2
DHT = dihydrotestosterone
ERE = estrogen response element
RING = A-ring or D-ring metabolic pathway
COMT= catechol-O-methyltransferase (COMT) gene
Clinical Message
The type, amount, and route of ET prescribed will be additive to that which is endogenously produced and will influence the hepatic synthesis of SHBG and therefore the bioavailability of the prescribed estrogen.
Estrogen Receptor Isoforms
The ERs are ligand-activated transcription factors. ERalpha and ERbeta have very similar DNA-binding domains, but they differ considerably in the N-terminal domain and to a lesser extent in the ligand-binding domain. As a result, various ligands have different binding affinities for the two receptors.[1,5,7] For example, if the relative binding affinity (RBA) of E2 is 100 for both receptors, the RBA of E1 is 60 for ERalpha and 37 for ERbeta.[9] Although both ERs are distributed throughout the body, ERalpha is expressed primarily in the uterus, liver, kidney, and heart, whereas ERbeta is expressed principally in the bladder and the central nervous system.[5] ERalpha and ERbeta are co-expressed in the breast, bone, and certain regions of the brain.[5] The estrogen-ER complexes bind to the estrogen response element as homodimers (ERalpha/ERalpha; ERbeta/ERbeta) or as heterodimers (ERalpha/ERbeta). ERbeta downregulates ERalpha-mediated transcriptional activity.[5]
Pharmacology: Not All Estrogens Are the Same
There is a paucity of validated clinically relevant data comparing and evaluating the pharmacokinetics and pharmacodynamics of CEE, E2, and EE. All of the preparations do elicit appropriate pharmacologic estrogen-like activity in various target organs, but the response varies.[10,11]
Chemical Structure
The estrogens in question are all steroidal compounds. Both CEE and E2 are classified as natural estrogens and EE as a synthetic estrogen.[11] In terms of human biology, only E2 should be regarded as natural, however, as both of the other estrogen compounds have components that are not found in women (pre- or postmenopausal) even though these components do have estrogen-like activity. Of the 2 steroidal estrogens, E2 may be further defined as a simple estrogen and CEE as a complex compound.
CEE has many components, some of which have not as yet been fully characterized.10 As shown in Table 1, 17alpha-dihydroequilin sulfate (15%) are the other major estrogens. Although certain of the estrogens are present in small quantities, their estrogen potency may be significant. For example, 17beta-dihydroequilin represents 1% to 2% of the CEE dose, but it is 8-fold more potent than E2 as an endometrial stimulant.[12] 17beta-dihydroequilin is not measurable in estradiol assays. In addition, 7 progestins and 4 androgens have been identified in CEE (Table 2 and Table 3).[11] CEE is available in oral tablets and in vaginal creams.
Table 1.
Conjugated Equine Estrogens: Estrogen Components[11]
| Estrogen* | Approximate Percentage of Total Dose |
|---|---|
| Estrone sulfate | 45 |
| Equilin sulfate | 25 |
| 17alpha-Dihydroequilin sulfate | 15 |
| delta 8, 9-Estrone sulfate | 3.5 |
| 17beta-Estradiol sulfate | 1-2 |
| 17alpha-Estradiol sulfate | 1-2 |
| 17beta-Dihydroequilin sulfate | 1-2 |
| Equilenin sulfate | 1-2 |
| 17beta-Dihydroequilenin sulfate | 1-2 |
| 17alpha-Dihydroequilenin sulfate | 1-2 |
| 17alpha-Dihydro-delta-estrone sulfate | ND |
| 17beta-Dihydro-delta-estrone sulfate | ND |
| 2-Hydroxyestrone sulfate | ND |
| 2-Methoxyestrone sulfate | ND |
| 5,7,9(10)-Estratriene-3beta, 17beta-diol sulfate | ND |
| 3beta-Hydroxy-5,7,9(10)-estratrien-17-one sulfate | ND |
| 3beta-Hydroxy-5(10),7-estradiene-17-one-3-sulfate (Estradiene) | ND |
Present as a sodium salt
ND = not determined
Table 2.
Conjugated Equine Estrogens: Identified Progestin Components[11]
| 5alpha-Pregnane-3beta, 20beta-diol-3-sulfate |
| 5alpha-Pregnane-3beta, 20beta-diol-20-sulfate |
| 3beta-Hydroxy-5alpha-pregnan-20-one |
| 5alpha-Pregnane-3beta, 20beta-diol-disulfate |
| 3beta-Hydroxy-5alpha-pregn-16-en-20-one |
| 5alpha-Pregnane-3beta, 16alpha, 20alpha-triol |
| 20alpha-Dihydro-4 pregnen-3-one-20-sulfate |
Table 3.
Conjugated Equine Estrogens: Identified Androgen Components[11]
| 3beta-Hydroxy-5alpha-androstan-16-one |
| 5alpha-Androstane-3beta, 16alpha-diol |
| 5alpha-Androstane-3beta, 16beta-diol |
| 5alpha-Androstane-3beta, 17beta-diol |
E2 is a single compound and is the major biologically active estrogen in humans. The E2 used in clinical practice is prepared from a nonestrogenic steroid precursor (diosgenin) found in yams and soy beans.[11] E2 is micronized to improve its absorption and is available in a variety of forms – tablets, rings, creams, patches and gels.
EE is synthesized from plant steroids and is biochemically similar to E2 (EE has an ethinyl group at carbon 17) but is much more potent than E2. EE is available only in an oral form (for ET) and is not measurable using routine estrogen assays. Other estrogens available for ET include esterified estrogen, synthetic conjugated estrogens, and piperazine estrone sulfate. Details about these estrogens are available elsewhere.[10]
Metabolism
The metabolism of orally administered estrogen is complex and subject to much inter- and intra-individual variability.[10,11] This is especially true for CEE because of its numerous components and more than 200 metabolites. Unconjugated estrogens (eg, equilin) are absorbed more rapidly than conjugated estrogens (eg, equilin sulfate), but they are soon conjugated by the liver (first-pass effect) and circulate together with E1S as a hormonally inert estrogen reservoir. Tissue enzymes, such as sulfatases and 17beta-hydroxysteroid dehydrogenases originating in bowel bacterial flora and the intestinal mucosa, activate these estrogens locally to E1 and E2.[11] The bioavailability of the estrogens is modulated by their binding to SHBG.
Only about 5% (range, 1% to 12%) of oral E2 is systemically bioavailable as a result of the entero-hepatic metabolism of E2 to E1 and its subsequent conversion and storage as E1S.[13] It has been estimated that 65% of E2 and 54% of E1 entering the circulation are converted to E1S. This is a reversible reaction, with 14% and 21% of E1S being converted to E1 and E2, respectively.[14] There is a sound teleological reason for this estrogen storage bank.[15] E2 has a rapid distribution phase of about 6 minutes and an elimination phase of only 2 hours. Tissues can thus theoretically draw on E1S (which has a plasma half-life of 5 to 9 hours) for bioavailable estrogen as needed. Serum E1 levels are higher than those of E2 after oral ET as a result of the hepatic 17beta-hydroxysteroid dehydrogenase type 2 activity that converts E2 to E1. The resulting increased E1/E2 ratio is similar to the physiologic change in the estrogen ratio that occurs postmenopausally.[11] The blood levels of E2, E1, and E1S may also depend on the route of ET administration. One small study found elevated levels of E1S after oral E2 as compared with transdermal E2.[13] Although comparable serum E2 levels were obtained after 7 months of treatment with 50 mcg transdermal E2 (70 pg/mL) and 1 mg oral E2 (72 pg/mL), the respective values for E1S were 2.3 ng/mL and 24.9 ng/mL.[13] (The normal E1S value during the menstrual cycle is 2 to 3 ng/mL.) In addition, a study has found that urinary excretion of the A- and D-ring metabolites 2-hydroxyestrone and 16alpha-hydroxyestrone, respectively, was higher for orally administered E2 compared with transdermal E2, although ratios of the A- to D-ring metabolites remained the same for both administration routes.[16] The clinical significance of these differences is not known.
Estrogen Transport and Bioavailability
About 1% of E2 circulates as a free steroid; 40% is bound with high affinity to SHBG, and the remainder is loosely attached to albumin.[11] Both the free and the albumin-bound E2 are bioavailable to various tissues. Although SHBG synthesis is genetically controlled, exogenous oral estrogens stimulate increased production of this protein (in the liver cells), whereas androgens suppress SHBG levels. Unconjugated equilin, E1, 17beta-dihydroequilin, and E2 bind with high but different affinities to SHBG. The differences reflect the potency of the various estrogens in terms of their stimulation of hepatic SHBG synthesis. Thus, in a comparative study, 0.625 mg CEE increased SHBG by 100% over baseline after 3 months of treatment; the increases in plasma SHBG after 1 mg of oral E2 and 50 mcg of transdermal E2 were 45% and 12%, respectively.[17] EE is the most potent estrogen in this regard, both in terms of stimulating the synthesis of SHBG as well as other hepatic proteins (eg, cortisol-binding protein; angiotensinogen) and suppressing follicle-stimulating hormone production.[10,11] On the basis of hepatic protein synthesis, 10 mcg of EE was shown to be equivalent in potency to 1.25 mg of CEE.[18] In one of the few head-to-head clinical studies that compared 2 doses of CEE (0.625mg and 1.25mg) and E2 (1 mg and 2 mg) on pituitary function, 1 mg of oral E2 reduced FSH to a lesser degree (34.6 mIU/mL) when compared with 0.625 mg CEE (50.3 mIU/mL) and the 2 higher doses of estrogen.[19] However, the estrogenicity of CEE relative to E2 may vary with a given component's potency in a particular target tissue. The CEE component, equilin sulfate, for example, is less effective than E2 in inhibiting bone resorption.[20]
Serum Estrogen Levels
Tables that compare serum E1 and E2 levels in postmenopausal women using various commercial products as an indication of their therapeutic equivalence are misleading.[11,21] E1 and E2 assays vary and do not measure potent estrogens such as EE and the CEE components. The duration of ET, the timing of sampling, and steady-state pharmacokinetics are additional important variables. Total E2 values are not reflective of free bioavailable estrogen: as mentioned, one study found that 1 mg of oral E2 and 50 mcg of transdermal E2 resulted in equivalent levels of total serum E2 – 72 and 70 pg/mL, respectively.[13] However, when free E2. is measured, 1 mg of oral E2 and 25 mcg of transdermal E2 are equivalent doses.[22] In addition, blood levels of estrogen do not reflect the metabolism of estrogen pro-hormones (E1S and E1) to bioavailable E2 by target tissue enzymes or quantify the local tissue effect of estrogen metabolites. This may be of particular importance in the breast and the potential for estrogen-associated breast cancer as well as for cardiovascular disease. In a study of healthy postmenopausal women, 2 mg of oral E2 markedly enhanced endothelium-dependant vasodilation, whereas 50 mcg/day of transdermal E2, which produced a similar increase in free E2, had no effect on vascular function.[22]
Hepatic Function
The type, dose, and route of ET influence aspects of hepatic function that may affect the risk for cardiovascular disease.[10] For example, whereas both EE and CEE increase triglyceride levels,[10] E2 (oral and transdermal) does not.[10] Oral estrogens (depending on the dose), but not equivalent doses of transdermal estrogen, increase certain procoagulant factors.[10] Higher doses of oral estrogen (CEE 1.25 mg) increase insulin resistance, whereas lower doses enhance insulin sensitivity.[10] Levels of inflammatory biomarkers and cytokines increase to a greater extent with oral vs transdermal estrogen. The clinical significance of these varied responses in healthy women is open to question. However, what these differences do illustrate is that all estrogens do not have the same quantitative and qualitative pharmacologic effect.
Estrogen Therapy : Clinical Application
Symptoms attributed to estrogen deficiency are the primary indication for ET, with the most prevalent being vasomotor symptoms (hot flashes) and urogenital atrophy.
Vasomotor Symptoms
Biologic Rationale
Although hot flashes are experienced by the majority of menopausal women, their response to estrogen deficiency and ET varies. For example, many perimenopausal menstruating women have estrogen-responsive hot flashes,[23] but hot flashes that are not estrogen responsive occur in 20% of naturally menopausal and 10% of surgically menopausal women – all of whom are estrogen deficient.[24] In addition, the intensity and frequency of this often debilitating symptom is variable, as is its duration. Hot flashes persist in 20% of women for up to 15 years postmenopause.
Estrogen deficiency has a central role in the pathogenesis of hot flashes, although various vasoactive and endocrine factors may be responsible for the heterogeneity in clinical presentation.[25] Other possible contributing factors include the following: a lower than normal hypothalamic thermoregulatory threshold to circulating levels of estrogen; varying sensitivity to fluctuating levels of estrogen; or a destabilized thermoregulatory set point resulting from an alteration in the ratio of serotonin (5-HT) receptors in favor of the hyperthermic 5-HT2a receptor. Estrogen modulates the activity of the 5-HT2a and the 5-HT1a (hypothermic) receptors, enhances serotonin synthesis, and increases the transport of serotonin into neurons.[25] Dehydroepiandrosterone sulfate (DHEA-S) – the adrenal androgen – correlates with vasomotor instability protection,[26] and high doses of oral estrogen decrease adrenal DHEA-S synthesis.[27]
Clinical Trials
A systematic review of 32 RCTs confirmed that CEE and E2 had significant and comparable effects on the treatment of menopausal hot flashes.[28] ET dosage is an important determinant of the speed of response, degree of efficacy, and unwanted side effects. Rapid reduction (within 2 to 4 weeks) of moderate to severe hot flashes was shown in an RCT to result with either 1 mg or 2 mg of oral E2.[29] It took 8 weeks to achieve a positive response with 0.5 mg of oral E2, and treatment with 0.25 mg of oral E2 for this duration yielded an inadequate response (ie, not different from placebo). Discontinuation rates of ET adverse events increased with dosage: 0.5 mg (5%), 1 mg (9%), 2 mg (16%).[29] Lower doses of CEE (0.45 mg and 0.3 mg) reduce hot flashes (compared with placebo), but not to the same degree as the standard dose of 0.625mg.[30] Addition of the progestin medroxyprogesterone acetate (MPA) to the various CEE doses did not change the hot flash response. Low doses of transdermal E2 (25 mcg and 37.5 mcg) are also effective in controlling hot flashes.[31] The dosage equivalent response of the various types and routes of ET is difficult to establish. Assessment of the responder analysis (a ≥ 90% reduction in the hot flash weekly weighted score) would be a more informative method of determining efficacy. Using this approach, for example, reduced the placebo effect in one study from 50% to < 15%.[29]
Clinical Practice
The lowest effective dose of ET should be prescribed consistent with the patient's total requirement for therapy. The duration of treatment should not be limited to an arbitrary period (eg, 5 years) but should be dictated by clinical needs and safety monitoring. The initial dose should be gradually titrated downwards and adjusted according to the response of the patient. Failure of hot flash symptoms to respond to an adequate dose of ET may be due to the type and/or route of ET. Increased SHBG binding and a decrease in bioavailable estrogen is often the reason for the lack of response. Changing the type of oral estrogen (eg, CEE to E2) or the route of ET (oral to transdermal) usually resolves the issue. Persistent hot flashes occasionally result from too high a dose of ET as a result of estrogen tachyphylaxis.[32]
Urogenital Atrophy
Biologic Rationale
The pathogenesis of urogenital atrophy (UGA) – atrophic vaginitis; urethral syndrome (urgency, nocturia without dysuria); urinary incontinence – is directly associated with the decreased synthesis and availability of estrogen.[33] The distribution and type of ER present in the pelvic tissues – vaginal epithelium, trigone of the bladder, urethra and supporting tissues of the pelvic floor – vary with reproductive status and age. For example, the number of ERbeta decreases significantly with menopause. Estrogen maintains the health of the vaginal epithelium and neighboring structures via 2 main physiologic pathways – pelvic blood flow and glycogenization of the vaginal epithelium–which lead to moisturization of the vagina, proliferation of protective colonies of lactobacilli, and the inhibition of E coli and growth of other gram-negative bacteria.[33] With regard to the urethra, estrogen increases the urethral closure pressure[34] and elevates the bladder's sensory threshold.[35] Estrogenization of the vaginal epithelium is easily assessed by the maturation index and by the vaginal pH. The latter is a simple and well-validated technique[36] and is also reflective of the pH changes in the proximal and distal urethra.[33] pH values in excess of 4.5 to 5.0 are indicative of atrophy.[36]
Clinical Trials
A number of clinical trials have consistently documented the efficacy of local ET for the treatment of atrophic vaginitis and the urethral syndrome. Equivalent results are obtained irrespective of the method of estrogen administration, ie, cream, tablets, or rings. However, some studies documented greater patient acceptance and compliance with tablets or rings.[37,38] The pharmacokinetics of estrogen absorption varies with the type of preparation used. Various estrogen creams may result in higher systemic estrogen levels and an increased incidence of endometrial hyperplasia.[38] This is mainly a dose-related phenomenon and can be avoided by the use of slow-release vaginal matrix E2 tablets. Use of the vaginal estrogen tablet restores vaginal and urethral cytology to normal, with systemic E2 values slightly elevated relative to baseline,[39] but within normal limits for postmenopausal women. Low-dose vaginal ET has been shown to effectively treat UGA in both short- and long-term studies. In one study, no endometrial hyperplasia developed after 1 year of use of the E2 vaginal matrix tablet.[40]
Nonetheless, a meta-analysis of RCTs evaluating systemic ET for the treatment of urinary incontinence reported mixed results.[41] Three recent, large RCTs concluded that postmenopausal hormone therapy (including oral and transdermal estrogen-alone treatment) increases the risk of developing urinary incontinence.[42–44] These results are counterintuitive, given the well-documented positive biologic effect of estrogen on the pelvic tissues. The role of ER heterogeneity and affinity for estrogen or estrogen-like ligands may provide the answer. It has been found, for example, that a selective estrogen receptor modulator (SERM) with some pelvic estrogen agonist activity (levormeloxifene) was associated with increased urinary incontinence,[45] whereas another SERM with limited estrogen activity in the reproductive tract (raloxifene) did not.[46] Systemic estrogen increases the synthesis of enzymes such as matrix metalloproteinases, which might theoretically degrade the collagen-rich pelvic ligament supports.
Clinical Practice
The true incidence of UGA is not known but approaches 40% of older women in some estimates.[33] Unfortunately, many women are hesitant to discuss symptoms associated with UGA, and many believe that incontinence is a normal consequence of aging. Notably, healthcare providers overestimate the estrogenization of the lower genital tract that can be achieved with systemic ET. In an oral estrogen dose-response study, the percentage of days without vaginal dryness in women taking placebo improved from a mean baseline of 60.5% to 73.8% at the end of the study, although the mean percentage of parabasal cells (indicative of vaginal atrophy) in the group increased from 8.7% at the start to 15.2% at the end of treatment.[47] An estimated 10% to 25% of women on adequate systemic hormone therapy have evidence of atrophic vaginitis.
Clinical Message
The health of the vaginal epithelium is best monitored by regular vaginal pH testing. Low-dose vaginal ET is best for UGA and can also be used to supplement systemic ET when indicated.
From Research to Clinical Practice
Advances in molecular and genomic science have confirmed that every woman is unique and has a thumbprint that will determine her response to physiologic events such as her menarche and menopause, to aging, and to intercurrent lifetime risks of disease. This includes her response to disease-specific therapies. Present technology does not permit the clinically practical use of this knowledge. The clinical trend is to simplify complex biology and, despite evidence to the contrary, to regard and treat women generically. Thus, all menopausal women cease to menstruate; most menopausal women have hot flashes, and over time may develop urogenital symptoms. Most will respond to a standard dose of estrogen. The efficacy and safety of ET is evaluated by RCTs with the assumption that (1) the women allocated to active drug and placebo groups are biologically the same and (2) the chosen ET and dose are appropriate for every woman in the study. The result of the study is deemed to be representative of an ET class effect and thus applicable to all menopausal women.
The endocrinology of the menopausal transition is complex. Variations in the genetically determined biology of women will determine the symptomatic response of a given menopausal woman to her physiologic decrease in estrogen synthesis and postmenopausal metabolism and to her response (or nonresponse) to ET. By definition all estrogens are the same. However, the source, chemical structure, and pharmacology of estrogens are different, and for a significant minority of women this factor will determine the clinical efficacy and safety of a particular ET for them. RCTs are not structured or designed to account for these individualized clinical variables.
The thoughtful extrapolation of clinical trial data can nevertheless avoid conflicting recommendations by ensuring applicability of a study result to a relevant population. For example, one trial concluded that HT does not have a meaningful effect on the health-related quality of life of postmenopausal women.[48] This conclusion is counterintuitive and contradicts the conclusion from another randomized trial[49] and clinical experience. Consider the difference between the 2 study populations: the former study[48] involved mainly asymptomatic women (mean age, 63 years) and the latter study[49] symptomatic perimenopausal and early postmenopausal women.
The translation of new information from the research bench to clinical trial and to the practice of medicine at a community level has been addressed by Lenfant.[50] In short, the author emphasizes that care must be taken to ensure that “when recommendations actually influence the way medicine is practiced,” so-called guidelines should not undermine “the role of independent judgment” since “the art of medicine will always be central to its responsible practice.”
Conclusion
I propose the following principles of management of symptomatic menopause with regard to ET[51]:
There must be a scientific rationale for use of ET.
The patient must have a valid indication for ET.
The type, dose, and route of ET must be individualized for a given indication at a given point in time.
The correct effective dose of hormone therapy (ie, estrogen or estrogen plus progestin) should be prescribed, reevaluated annually, and adjusted to the woman's clinical response and total needs. There is no arbitrary time limit to the indication for ET.
There is no single preparation of hormone therapy that is suitable for all women.
Funding Information
This article was supported in part by an independent and unrestricted educational grant from Novo Nordisk.
References
- 1.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med. 2002;346:340–352. doi: 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- 2.Weiss G, Skurnick JH, Goldsmith LT, et al. Menopause and hypothalamic-pituitary sensitivity to estrogen. JAMA. 2004;292:2991–2996. doi: 10.1001/jama.292.24.2991. [DOI] [PubMed] [Google Scholar]
- 3.Women's Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy. The Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 4.Notelovitz M. Age-adjusted long-term estrogen therapy: biologic and pharmacologic principles for clinical practice. Medscape General Medicine. 2006 [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsson S, Makela S, Treuter E, et al. Mechanism of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 6.Levin ER. Cellular functions of plasma membrane estrogen receptors. Steroids. 2002;67:471–475. doi: 10.1016/s0039-128x(01)00179-9. [DOI] [PubMed] [Google Scholar]
- 7.Clemmons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001;344:276–285. doi: 10.1056/NEJM200101253440407. [DOI] [PubMed] [Google Scholar]
- 8.Labrie F, Belanger A, Luu-The V, et al. DHEA and intracrine formation of androgens and estrogens in peripheral target tissues: its role during aging. Steroids. 1998;63:322–328. doi: 10.1016/s0039-128x(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 9.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19:1–27. doi: 10.1093/carcin/19.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Ansbacher R. The pharmacokinetics and efficacy of different estrogens are not equivalent. Am J Obstet Gynecol. 2001;184:255–263. doi: 10.1067/mob.2001.109656. [DOI] [PubMed] [Google Scholar]
- 11.Stanczyk F. Estrogens used for replacement therapy in postmenopausal women. Gynecol Endocrinol. 2001;15(suppl 4):17–25. [Google Scholar]
- 12.Bhavnani BR. Pharmacokinetics and pharmacodynamics of conjugated equine estrogens: chemistry and metabolism. Proc Soc Exp Biol Med. 1998;217:6–16. doi: 10.3181/00379727-217-44199. [DOI] [PubMed] [Google Scholar]
- 13.Slater CC, Hodis HN, Mack WJ, et al. Markedly elevated levels of estrone sulfate after long-term oral, but not transdermal, administration of estradiol in postmenopausal women. Menopause. 2001;8:200–203. doi: 10.1097/00042192-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 14.Buster JE. Estrogen kinetics for clinicians. In: Sciarra JJ, editor. Gynecology and Obstetrics: Endocrinology,Infertility, Genetics. Vol. 5. Philadelphia, Pa: JB Lippincott; 1994. pp. 1–16. [Google Scholar]
- 15.Roberts KD, Rochefort JG, Bleu G, Chapdelaine A. Plasma estrone sulfate levels in postmenopausal women. Steroids. 1980;35:179–187. doi: 10.1016/0039-128x(80)90101-4. [DOI] [PubMed] [Google Scholar]
- 16.Lippert TH, Seeger H, Mueck AO. Estradiol metabolism during oral and transdermal estradiol replacement therapy in postmenopausal women. Horm Metab Res. 1998;30:598–600. doi: 10.1055/s-2007-978940. [DOI] [PubMed] [Google Scholar]
- 17.Nachtigall LE, Raju U, Banerjee S, et al. Serum estradiol-binding profiles in postmenopausal women undergoing three common estrogen replacement therapies: associations with sex hormone-binding globulin, estradiol and estrone levels. Menopause. 2000;7:243–250. doi: 10.1097/00042192-200007040-00006. [DOI] [PubMed] [Google Scholar]
- 18.Mandel FP, Geola FL, Lu JK, et al. Biologic effects of various doses of ethinyl estradiol in postmenopausal women. Obstet Gynecol. 1982;59:673–679. [PubMed] [Google Scholar]
- 19.Archer DF, Fischer LA, Rich D, et al. Estrace vs Premarin for treatment of menopausal symptoms: dosage comparison study. Adv Ther. 1992;9:21–31. [Google Scholar]
- 20.Lobo RA, Nguyen HN, Eggena P, Brenner PF. Biologic effects of equilin sulfate in postmenopausal women. Fertil Steril. 1988;49:234–238. doi: 10.1016/s0015-0282(16)59708-8. [DOI] [PubMed] [Google Scholar]
- 21.Boothby LA, Doering PL, Kipersztok S. Bioidentical hormone therapy: a review. Menopause. 2004;11:356–367. doi: 10.1097/01.gme.0000094356.92081.ef. [DOI] [PubMed] [Google Scholar]
- 22.Vehkavaara S, Hakala-Ala-Pietila T, Virkamaki A, et al. Differential effects of oral and transdermal estrogen replacement therapy on endothelial function in postmenopausal women. Circulation. 2000;102:2687–2693. doi: 10.1161/01.cir.102.22.2687. [DOI] [PubMed] [Google Scholar]
- 23.Oldenhave A, Jaszmann IJ, Haspels AA, et al. Impact of climacteric on well-being. A survey based on 5313 women 39 to 60 years old. Am J Obstet Gynecol. 1993;168:772–780. doi: 10.1016/s0002-9378(12)90817-0. [DOI] [PubMed] [Google Scholar]
- 24.Bachmann GA. Vasomotor flushes in menopausal women. Am J Obstet Gynecol. 1999;180:312–316. doi: 10.1016/s0002-9378(99)70725-8. [DOI] [PubMed] [Google Scholar]
- 25.Stearns V, Ullmer L, Lopez JF, et al. Hot flushes. Lancet. 2002;360:1851–1861. doi: 10.1016/s0140-6736(02)11774-0. [DOI] [PubMed] [Google Scholar]
- 26.Overlie I, Moen MH, Holte A, Finset A. Androgens and estrogens in relation to hot flushes during the menopausal transition. Maturitas. 2002;41:69–77. doi: 10.1016/s0378-5122(01)00256-0. [DOI] [PubMed] [Google Scholar]
- 27.Casson PR, Elkind-Hirsch KE, Buster JE, et al. Effect of postmenopausal estrogen replacement on circulating androgens. Obstet Gynecol. 1997;90:995–998. doi: 10.1016/s0029-7844(97)00538-3. [DOI] [PubMed] [Google Scholar]
- 28.Nelson HD. Commonly used types of postmenopausal estrogen for treatment of hot flashes: scientific review. JAMA. 2004;291:1610–1620. doi: 10.1001/jama.291.13.1610. [DOI] [PubMed] [Google Scholar]
- 29.Notelovitz M, Lenihan JP, McDermott M, et al. Initial 17beta estradiol dose for treating vasomotor symptoms. Obstet Gynecol. 2000;95:726–731. doi: 10.1016/s0029-7844(99)00643-2. [DOI] [PubMed] [Google Scholar]
- 30.Utian WH, Shoupe D, Bachmann GA, et al. Relief of vasomotor symptoms and vaginal atrophy with lower doses of conjugated equine estrogens and medroxyprogesterone acetate. Fertil Steril. 2001;75:1065–1079. doi: 10.1016/s0015-0282(01)01791-5. [DOI] [PubMed] [Google Scholar]
- 31.Utian WH, Burry KA, Archer DF, et al. Efficacy and safety of low, standard and high dosages of an estradiol transport system (Esclim) compared with placebo on vasomotor symptoms in highly symptomatic menopausal patients. Am J Obstet Gynecol. 1999;181:71–79. doi: 10.1016/s0002-9378(99)70438-2. [DOI] [PubMed] [Google Scholar]
- 32.Gangar K, Cust M, Whitehead MJ. Symptoms of oestrogen deficiency associated with supraphysiological plasma oestradiol concentrations in women with oestradiol implants. BMJ. 1989;299:601–602. doi: 10.1136/bmj.299.6699.601-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Notelovitz M. Urogenital atrophy and low-dose vaginal estrogen therapy. Menopause. 2000;7:140–142. [PubMed] [Google Scholar]
- 34.Bhatia NN, Bergman A, Karram MM. Effects of estrogen on urethral function in women with urinary incontinence. Am J Obstet Gynecol. 1989;160:176–181. doi: 10.1016/0002-9378(89)90114-2. [DOI] [PubMed] [Google Scholar]
- 35.Fantl JA, Wyman JF, Anderson RL, et al. Postmenopausal urinary incontinence: comparison between non-estrogen supplemented and estrogen supplemented women. Obstet Gynecol. 1988;71:823–828. [PubMed] [Google Scholar]
- 36.Brizzolara S, Killeen J, Severino R. Vaginal pH and parabasal cells in postmenopausal women. Obstet Gynecol. 1999;94:700–703. doi: 10.1016/s0029-7844(99)00384-1. [DOI] [PubMed] [Google Scholar]
- 37.Bachmann G, Notelovitz M, Nachtigall L, Bergerson L. A comparative study of low-dose estradiol vaginal ring and conjugated estrogen cream for postmenopausal urogenital atrophy. Prim Care Update Ob/Gyns. 1997;4:109–115. [Google Scholar]
- 38.Rioux JE, Devlin MC, Gelfand MM, et al. 17beta estradiol vaginal tablet versus conjugated equine estrogen vaginal cream to relieve menopausal atrophic vaginitis. Menopause. 2000;7:156–161. doi: 10.1097/00042192-200007030-00005. [DOI] [PubMed] [Google Scholar]
- 39.Notelovitz M, Funk S, Nanavati N, Mazzeo M. Estradiol absorption from vaginal tablets in postmenopausal women. Obstet Gynecol. 2002;99:556–562. doi: 10.1016/s0029-7844(01)01385-0. [DOI] [PubMed] [Google Scholar]
- 40.Simunic V, Banovic J, Ciglar S, et al. Local estrogen treatment in patients with urogenital symptoms. Int J Gynaecol Obstet. 2003;82:187–197. doi: 10.1016/s0020-7292(03)00200-5. [DOI] [PubMed] [Google Scholar]
- 41.Fantl JA, Cardozo L, McClish DK. Estrogen therapy in the management of urinary incontinence in postmenopausal women: a meta-analysis. First report of the Hormones and Urogenital Committee. Obstet Gynecol. 1994;83:12–18. [PubMed] [Google Scholar]
- 42.Grady D, Brown JS, Vittinghoff E, et al. Postmenopausal hormones and incontinence: the Heart and Estrogen/progestin Replacement Study. Obstet Gynecol. 2001;97:116–120. doi: 10.1016/s0029-7844(00)01115-7. [DOI] [PubMed] [Google Scholar]
- 43.Jackson S, Shepherd A, Brookes S, Abrams P. The effect of estrogen supplementation on postmenopausal urinary stress incontinence: a double blind placebo controlled trial. Br J Obstet Gynaecol. 1999;106:711–718. doi: 10.1111/j.1471-0528.1999.tb08372.x. [DOI] [PubMed] [Google Scholar]
- 44.Grodstein F, Lifford K, Resnick NM, Curhan GC. Postmenopausal hormone therapy and risk of developing urinary incontinence. Obstet Gynecol. 2004;103:254–260. doi: 10.1097/01.AOG.0000107290.33034.6f. [DOI] [PubMed] [Google Scholar]
- 45.Goldstein SR, Nanavati N. Adverse events that are associated with the selective estrogen receptor modulator levormeloxifene in an aborted phase III osteoporosis treatment study. Am J Obstet Gynecol. 2002;187:521–527. doi: 10.1067/mob.2002.123938. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein SR. An update on non-uterine gynecological effects of raloxifene. Eur J Cancer. 2002;38(suppl 6):S65–S66. doi: 10.1016/s0959-8049(02)00291-5. [DOI] [PubMed] [Google Scholar]
- 47.Notelovitz M, Mattox JH. Suppression of vasomotor and vulvovaginal symptoms with continuous oral 17beta estradiol. Menopause. 2000;7:310–317. doi: 10.1097/00042192-200007050-00005. [DOI] [PubMed] [Google Scholar]
- 48.Hays J, Ockene JK, Brunner RL, et al. Effects of estrogen plus progestin on health related quality of life. N Engl J Med. 2003;348:1839–1854. doi: 10.1056/NEJMoa030311. [DOI] [PubMed] [Google Scholar]
- 49.Gambacciani M, Ciaponi M, Cappagli B, et al. Effects of low-dose, continuous combined estradiol and norethisterone acetate on menopausal quality of life in early postmenopausal women. Maturitas. 2003;44:157–163. doi: 10.1016/s0378-5122(02)00327-4. [DOI] [PubMed] [Google Scholar]
- 50.Lenfant C. Clinical research to clinical practice - lost in translation? N Engl J Med. 2003;349:868–874. doi: 10.1056/NEJMsa035507. [DOI] [PubMed] [Google Scholar]
- 51.Notelovitz M. The clinical practice impact of the Women's Health Initiative: political vs biologic correctness. Maturitas. 2003;44:3–9. doi: 10.1016/s0378-5122(02)00333-x. [DOI] [PubMed] [Google Scholar]