

Abstract
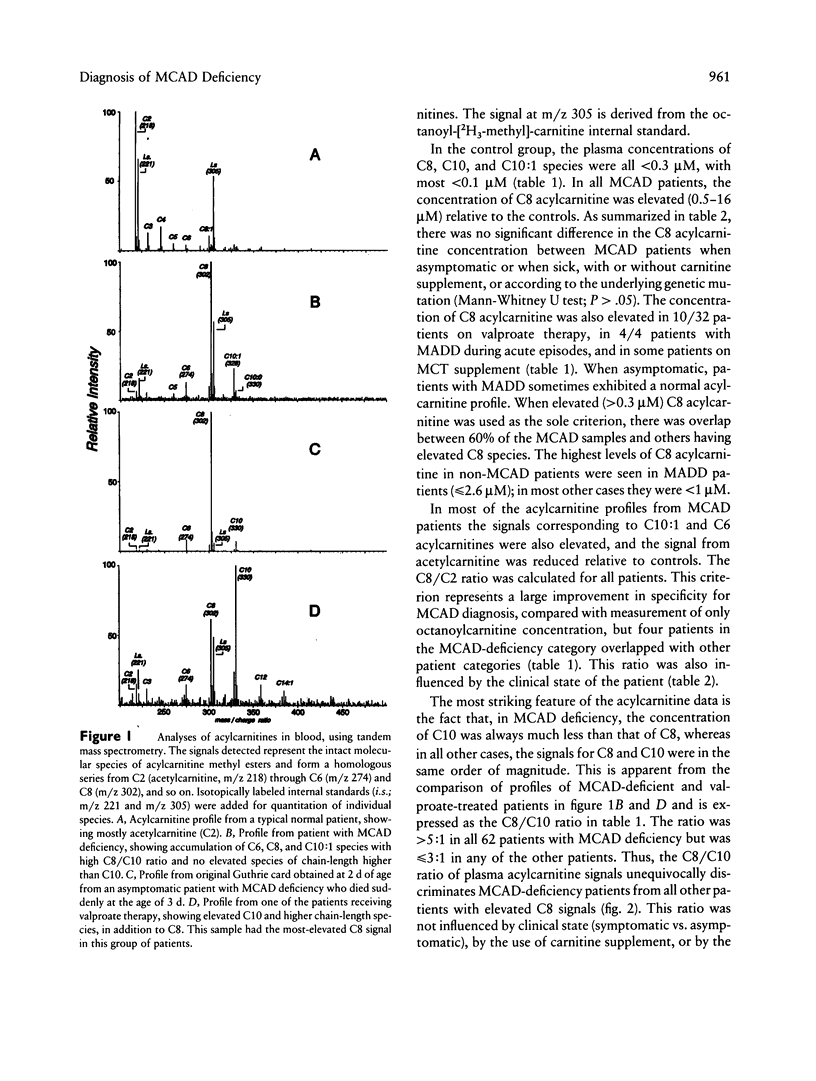
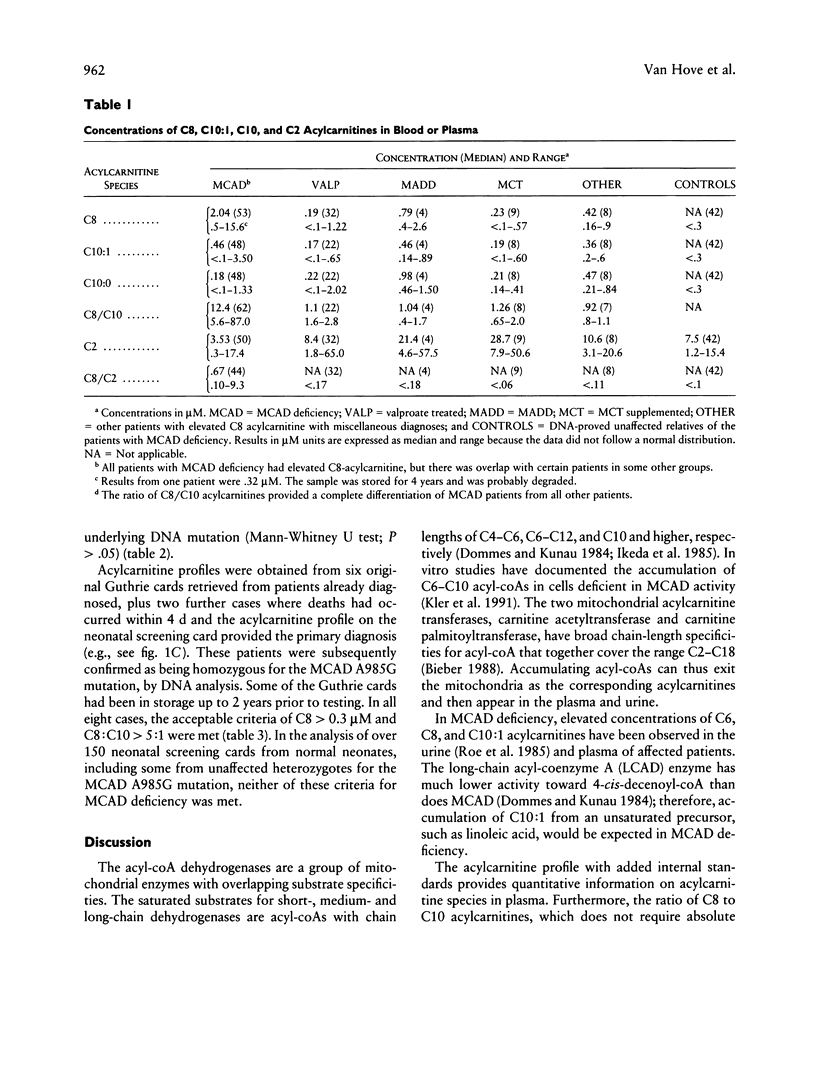
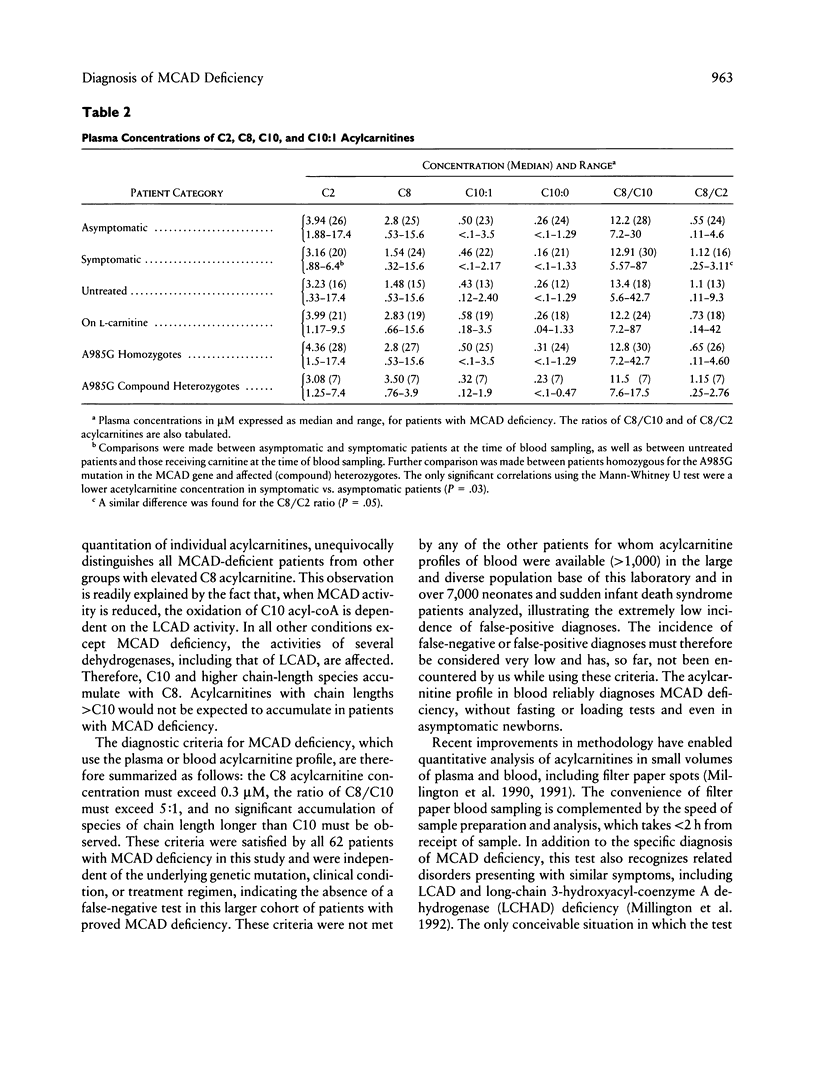
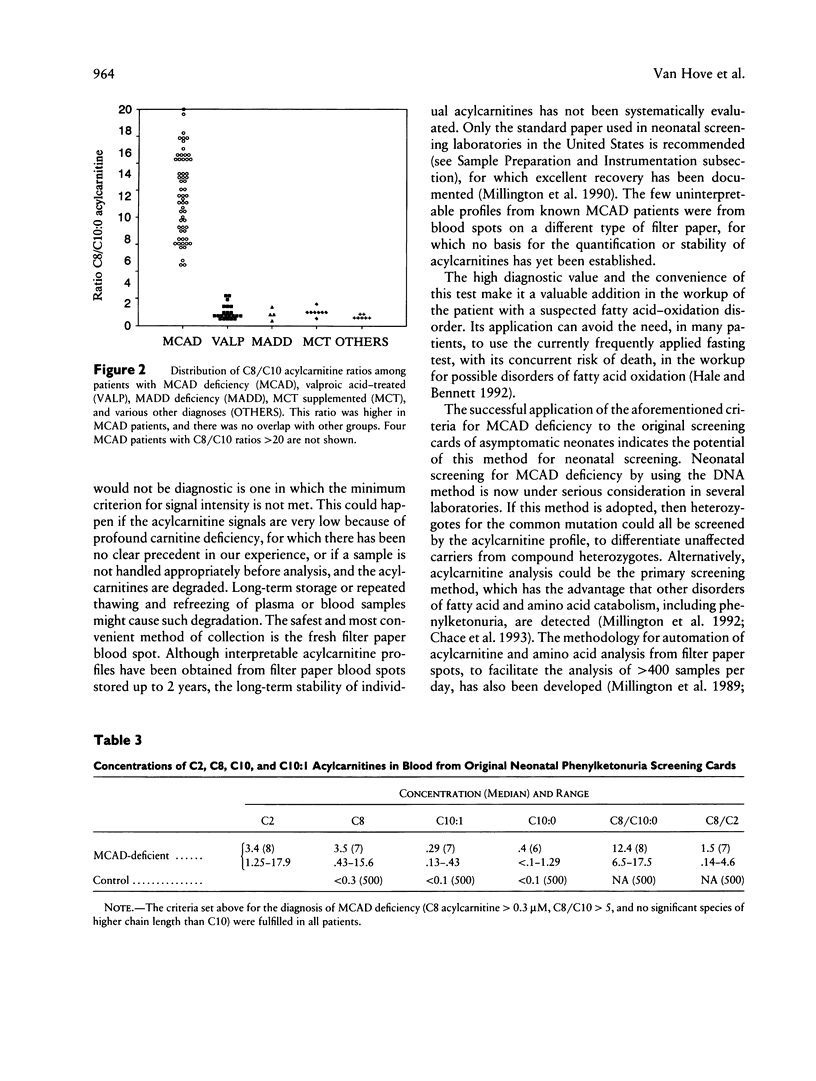
Medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency is a disorder of fatty acid catabolism, with autosomal recessive inheritance. The disease is characterized by episodic illness associated with potentially fatal hypoglycemia and has a relatively high frequency. A rapid and reliable method for the diagnosis of MCAD deficiency is highly desirable. Analysis of specific acylcarnitines was performed by isotope-dilution tandem mass spectrometry on plasma or whole blood samples from 62 patients with MCAD deficiency. Acylcarnitines were also analyzed in 42 unaffected relatives of patients with MCAD deficiency and in other groups of patients having elevated plasma C8 acylcarnitine, consisting of 32 receiving valproic acid, 9 receiving medium-chain triglyceride supplement, 4 having multiple acyl-coenzyme A dehydrogenase deficiency, and 8 others with various etiologies. Criteria for the unequivocal diagnosis of MCAD deficiency by acylcarnitine analysis are an elevated C8-acylcarnitine concentration (> 0.3 microM), a ratio of C8/C10 acylcarnitines of > 5, and lack of elevated species of chain length > C10. These criteria were not influenced by clinical state, carnitine treatment, or underlying genetic mutation, and no false-positive or false-negative results were obtained. The same criteria were also successfully applied to profiles from neonatal blood spots retrieved from the original Guthrie cards of eight patients. Diagnosis of MCAD deficiency can therefore be made reliably through the analysis of acylcarnitines in blood, including presymptomatic neonatal recognition. Tandem mass spectrometry is a convenient method for fast and accurate determination of all relevant acylcarnitine species.
Full text
PDF




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bieber L. L. Carnitine. Annu Rev Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- Blakemore A. I., Singleton H., Pollitt R. J., Engel P. C., Kolvraa S., Gregersen N., Curtis D. Frequency of the G985 MCAD mutation in the general population. Lancet. 1991 Feb 2;337(8736):298–299. doi: 10.1016/0140-6736(91)90907-7. [DOI] [PubMed] [Google Scholar]
- Catzeflis C., Bachmann C., Hale D. E., Coates P. M., Wiesmann U., Colombo J. P., Joris F., Délèze G. Early diagnosis and treatment of neonatal medium-chain acyl-CoA dehydrogenase deficiency: report of two siblings. Eur J Pediatr. 1990 May;149(8):577–581. doi: 10.1007/BF01957697. [DOI] [PubMed] [Google Scholar]
- Chace D. H., Millington D. S., Terada N., Kahler S. G., Roe C. R., Hofman L. F. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem. 1993 Jan;39(1):66–71. [PubMed] [Google Scholar]
- Coates P. M., Hale D. E., Stanley C. A., Corkey B. E., Cortner J. A. Genetic deficiency of medium-chain acyl coenzyme A dehydrogenase: studies in cultured skin fibroblasts and peripheral mononuclear leukocytes. Pediatr Res. 1985 Jul;19(7):671–676. doi: 10.1203/00006450-198507000-00007. [DOI] [PubMed] [Google Scholar]
- Ding J. H., Bross P., Yang B. Z., Iafolla A. K., Millington D. S., Roe C. R., Gregersen N., Chen Y. T. Genetic heterogeneity in MCAD deficiency: frequency of K329E allele and identification of three additional mutant alleles. Prog Clin Biol Res. 1992;375:479–488. [PubMed] [Google Scholar]
- Dommes V., Kunau W. H. Purification and properties of acyl coenzyme A dehydrogenases from bovine liver. Formation of 2-trans,4-cis-decadienoyl coenzyme A. J Biol Chem. 1984 Feb 10;259(3):1789–1797. [PubMed] [Google Scholar]
- Duran M., Bruinvis L., Ketting D., de Klerk J. B., Wadman S. K. Cis-4-decenoic acid in plasma: a characteristic metabolite in medium-chain acyl-CoA dehydrogenase deficiency. Clin Chem. 1988 Mar;34(3):548–551. [PubMed] [Google Scholar]
- Hale D. E., Bennett M. J. Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr. 1992 Jul;121(1):1–11. doi: 10.1016/s0022-3476(05)82532-6. [DOI] [PubMed] [Google Scholar]
- Heales S. J., Leonard J. V. Diagnosis of medium chain acyl CoA dehydrogenase deficiency by measurement of cis-4-decenoic acid in dried blood spots. Clin Chim Acta. 1992 Jul 31;209(1-2):61–66. doi: 10.1016/0009-8981(92)90333-l. [DOI] [PubMed] [Google Scholar]
- Heales S. J., Woolf D. A., Robinson P., Leonard J. V. Rapid diagnosis of medium-chain acyl CoA dehydrogenase deficiency by measurement of cis-4-decenoic acid in plasma. J Inherit Metab Dis. 1991;14(5):661–667. doi: 10.1007/BF01799930. [DOI] [PubMed] [Google Scholar]
- Ikeda Y., Okamura-Ikeda K., Tanaka K. Purification and characterization of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. Isolation of the holo- and apoenzymes and conversion of the apoenzyme to the holoenzyme. J Biol Chem. 1985 Jan 25;260(2):1311–1325. [PubMed] [Google Scholar]
- Kler R. S., Jackson S., Bartlett K., Bindoff L. A., Eaton S., Pourfarzam M., Frerman F. E., Goodman S. I., Watmough N. J., Turnbull D. M. Quantitation of acyl-CoA and acylcarnitine esters accumulated during abnormal mitochondrial fatty acid oxidation. J Biol Chem. 1991 Dec 5;266(34):22932–22938. [PubMed] [Google Scholar]
- Matsubara Y., Narisawa K., Miyabayashi S., Tada K., Coates P. M. Molecular lesion in patients with medium-chain acyl-CoA dehydrogenase deficiency. Lancet. 1990 Jun 30;335(8705):1589–1589. doi: 10.1016/0140-6736(90)91413-5. [DOI] [PubMed] [Google Scholar]
- Matsubara Y., Narisawa K., Tada K., Ikeda H., Yao Y. Q., Danks D. M., Green A., McCabe E. R. Prevalence of K329E mutation in medium-chain acyl-CoA dehydrogenase gene determined from Guthrie cards. Lancet. 1991 Aug 31;338(8766):552–553. doi: 10.1016/0140-6736(91)91110-g. [DOI] [PubMed] [Google Scholar]
- Millington D. S., Kodo N., Norwood D. L., Roe C. R. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321–324. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- Millington D. S., Norwood D. L., Kodo N., Roe C. R., Inoue F. Application of fast atom bombardment with tandem mass spectrometry and liquid chromatography/mass spectrometry to the analysis of acylcarnitines in human urine, blood, and tissue. Anal Biochem. 1989 Aug 1;180(2):331–339. doi: 10.1016/0003-2697(89)90441-7. [DOI] [PubMed] [Google Scholar]
- Millington D. S., Terada N., Chace D. H., Chen Y. T., Ding J. H., Kodo N., Roe C. R. The role of tandem mass spectrometry in the diagnosis of fatty acid oxidation disorders. Prog Clin Biol Res. 1992;375:339–354. [PubMed] [Google Scholar]
- Moon A., Rhead W. J. Complementation analysis of fatty acid oxidation disorders. J Clin Invest. 1987 Jan;79(1):59–64. doi: 10.1172/JCI112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton D. H., Kelley R. I. Diagnosis of medium-chain acyl-coenzyme A dehydrogenase deficiency in the neonatal period by measurement of medium-chain fatty acids in plasma and filter paper blood samples. J Pediatr. 1990 Sep;117(3):439–442. doi: 10.1016/s0022-3476(05)81091-1. [DOI] [PubMed] [Google Scholar]
- Rinaldo P., O'Shea J. J., Welch R. D., Tanaka K. Stable isotope dilution analysis of n-hexanoylglycine, 3-phenylpropionylglycine and suberylglycine in human urine using chemical ionization gas chromatography/mass spectrometry selected ion monitoring. Biomed Environ Mass Spectrom. 1989 Jul;18(7):471–477. doi: 10.1002/bms.1200180705. [DOI] [PubMed] [Google Scholar]
- Roe C. R., Millington D. S., Kahler S. G., Kodo N., Norwood D. L. Carnitine homeostasis in the organic acidurias. Prog Clin Biol Res. 1990;321:383–402. [PubMed] [Google Scholar]
- Roe C. R., Millington D. S., Maltby D. A., Bohan T. P., Kahler S. G., Chalmers R. A. Diagnostic and therapeutic implications of medium-chain acylcarnitines in the medium-chain acyl-coA dehydrogenase deficiency. Pediatr Res. 1985 May;19(5):459–466. doi: 10.1203/00006450-198505000-00011. [DOI] [PubMed] [Google Scholar]
- Roe C. R., Millington D. S., Maltby D. A., Kinnebrew P. Recognition of medium-chain acyl-CoA dehydrogenase deficiency in asymptomatic siblings of children dying of sudden infant death or Reye-like syndromes. J Pediatr. 1986 Jan;108(1):13–18. doi: 10.1016/s0022-3476(86)80762-4. [DOI] [PubMed] [Google Scholar]
- Schmidt-Sommerfeld E., Penn D., Duran M., Rinaldo P., Bennett M. J., Santer R., Stanley C. A. Detection and quantitation of acylcarnitines in plasma and blood spots from patients with inborn errors of fatty acid oxidation. Prog Clin Biol Res. 1992;375:355–362. [PubMed] [Google Scholar]
- Seakins J. W., Rumsby G. The use of phenylpropionic acid as a loading test for medium-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 1988;11 (Suppl 2):221–224. doi: 10.1007/BF01804241. [DOI] [PubMed] [Google Scholar]
- Stanley C. A., Hale D. E., Coates P. M., Hall C. L., Corkey B. E., Yang W., Kelley R. I., Gonzales E. L., Williamson J. R., Baker L. Medium-chain acyl-CoA dehydrogenase deficiency in children with non-ketotic hypoglycemia and low carnitine levels. Pediatr Res. 1983 Nov;17(11):877–884. doi: 10.1203/00006450-198311000-00008. [DOI] [PubMed] [Google Scholar]
- Yokota I., Coates P. M., Hale D. E., Rinaldo P., Tanaka K. Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet. 1991 Dec;49(6):1280–1291. [PMC free article] [PubMed] [Google Scholar]