

Abstract
Protein tyrosine phosphorylation accompanies and is essential for integrin signaling. We have shown that tyrosine phosphorylation of paxillin α and Crk-associated substrate (p130Cas) is a prominent event on integrin activation in normal murine mammary gland epithelial cells. Tyrosine phosphorylation of p130Cas has been demonstrated to facilitate cell migration. We show here that tyrosine phosphorylation of paxillin α acts to reduce haptotactic cell migrations as well as transcellular invasive activities in several different experimental cell systems, whereas tyrosine phosphorylation of p130Cas exerts opposing effects to those of paxillin α. Each of the phosphorylation-null mutants acts as a dominant negative for each phenotype. Moreover, we found that overexpression of paxillin α reduced the cell saturation density of normal murine mammary gland cells, whereas overexpression of p130Cas increased it. These effects also seemed to depend on tyrosine phosphorylation events. Cell growth rates and morphologies at growing phases were not significantly altered, nor were cells transformed. Addition of epidermal growth factor increased saturation density of the paxillin α-overexpressing cells, whereas no further increment was observed in p130Cas-overexpressing cells. We propose that tyrosine phosphorylation of paxillin α and p130Cas exerts opposing effects on several integrin-mediated cellular events, possibly through different signaling pathways.
Cell migration plays an essential role in a wide variety of physiological and pathological aspects of the organization of multicellular organisms. Cell migration is primarily mediated by integrin binding to extracellular matrices (ECMs) (reviewed in refs. 1–3). Integrins themselves have no intrinsic enzymatic activity, and a number of different cytoplasmic proteins, with scaffolding as well as signaling properties, must assemble on the cytoplasmic tails of integrins for proper functioning. Greater understanding of intracellular signaling through integrins reveals that highly complexed pathways operate to regulate cell motility. Moreover, cell motile activity is closely related to other cellular events such as cell morphology, growth, differentiation, and survival, in which a variety of different signals from other cell surface receptors are also involved. It is thus believed, but remains to be established, that some mechanism may exist that orchestrates and coordinates these distinct and diverse signals, culminating in cell migration.
One of the early cellular events that occurs on integrin activation is protein tyrosine phosphorylation. By use of epithelial–mesenchymal transdifferentiation (EMT) and migrating epithelial cells, we have shown that tyrosine phosphorylation of paxillin α and Crk-associated substrate (p130Cas) is a prominent intracellular event during integrin activation in normal murine mammary gland (NMuMG) epithelial cells (4). Protein levels of paxillin and p130Cas as well as tyrosine phosphorylation of other focal adhesion proteins such as focal adhesion kinase (Fak) were almost unchanged during EMT or epithelial cell migration.
p130Cas acts as an adaptor molecule in integrin signaling. p130Cas contains a tyrosine kinase substrate domain consisting of 15 potential src homology domain 2 (SH2)-binding motifs (5), nine of which conform to the SH2-binding motif for Crk [Tyr-Asp-(Val/Thr)-Pro] (6). Cell adhesion to ECM promotes Fak and c-Src kinase activity leading to tyrosine phosphorylation of p130Cas and its association with Crk and Nck (7, 8). Cary et al. (9) demonstrated that p130Cas acts as a mediator of Fak-promoted cell migration in Chinese hamster ovary cells (9). Assembly of p130Cas and Crk has been proposed to act as a molecular switch for induction of cell migration in FG-M carcinoma cells or in COS7 cells (10). p130Cas and the Crk complex then seem to lead to signaling to Rac activation, but not to Ras (10) or to Akt kinase (11). Similary, p130Cas signaling evoked by Fak does not seem to lead to extracellular signal-regulated protein kinase 1/2 activation (9).
Paxillin also acts as a scaffolding adaptor protein in integrin signaling by binding to several other integrin-assembly proteins, including vinculin, Fak, and c-Src (12, 13). Integrin-mediated tyrosine phosphorylation of paxillin also creates binding sites for several SH2-containing proteins such as Crk-I, Crk-II, Crk-L, and Csk (13). Tyrosine phosphorylation of paxillin has been shown to be important for cell cycle progression (14, 15) as well as adhesion-dependent function of leukocytes (16). Several tyrosine kinases, including Fak and Src-family kinases, have been implicated in paxillin phosphorylation (15, 17, 18). However, a precise mechanism for, as well as the role of tyrosine phosphorylation of paxillin during, cell migration remains to be established.
We here examined the role of tyrosine phosphorylation of paxillin α, in comparison with that of p130Cas, in integrin-mediated cellular events. Our results revealed that tyrosine phosphorylation of paxillin α and p130Cas opposingly regulate the cellular activity of directed migration along a gradient of immobilized factors (so-called haptotactic activity) of COS7 and NMuMG cells and transcellular invasive activities of MM1 hepatoma cells penetrating through mesothelial cell monolayers. We also revealed these proteins exert opposing effects on contact inhibition of growth of NMuMG cells. Possible downstream effectors of paxillin α and p130Cas were also explored.
Materials and Methods
Cell Culture.
NMuMG cells (CRL 1639) with a passage number of 15 were obtained from American Type Culture Collection and were grown or transdifferentiated by transforming growth factor β (TGF-β), as described (19). MM1 cells (20), COS7 cells (21), and mesothelial cells isolated from Donryu rat mesentery (20, 22) were grown as described previously.
cDNA Clones and Expressions.
Enhanced green fluorescent protein (EGFP)-tagged paxillin α cDNA in pBabePuro or in pEGFP-C1 (23), and the tyrosine phosphorylation-null mutant (the 4X) in which all four tyrosine phosphorylation sites (tyrosines 31, 40, 118, and 181) were mutated to phenylalanine were described previously (4). p EF-Bos GST (pEBG)/p130Cas and pEBG/p130CasΔSD (24) were gifts from H. Hirai (Tokyo University, Tokyo). cDNAs encoding EGFP-tagged p130Cas or phosphorylation-null mutants of p130Cas (the ΔSD mutant) were constructed by ligating the blunt-ended NotI-BamHI fragments, from pEBG/p130Cas or from its ΔSD mutant into BglII-SmaI sites of pEGFP-C1 (CLONTECH). Eco47 III-BamHI fragments were then isolated from each construct, blunt ended, and ligated into the SnaBI site of pBabePuro. pBabe plasmids were packaged into BOSC 23-derived retroviruses, as described (25). Virus titers were in the range of 105 − 106 infectious units per milliliter. NMuMG cells were infected with BOSC23 retroviruses and selected with 1 μg/ml puromycin (Sigma). Cells were then frozen in aliquots and in each experiment, cells were cultured for no longer than 2 weeks. COS7 cells (2 × 105 cells/6-cm dish) were transfected with each 2.6 μg of pEGFP-C1/paxillin α, the 4X mutant, pEGFP-C1/p130Cas, or the ΔSD mutant by using FuGENE6 (Boehringer–Mannheim) as described previously (21), with routine transfection efficiency of 40–60%. MM-1 cells were stably transfected with cDNAs in pEGFP-C1 by using electroporation as previously described (26).
Antibodies.
Antipaxillin antibody (Ab199–217) was described previously (23). Anti-p130Cas antibody was a gift from H. Hirai or purchased from Upstate Biotechnology. Antibodies against paxillin and Crk (Transduction Laboratories, Lexington, KY), Fak and phosphotyrosine (clone 4G10) (Upstate Biotechnology), CrkII, Crk L, Abl and Cbl (Santa Cruz Biotechnology), BrdUrd (Sigma), and horseradish peroxidase- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch) were purchased from commercial sources.
Cell Adhesion and Haptotactic Migration Assays.
Cell migration assays were performed by using modified Boyden chambers (tissue culture-treated, 6.5-mm diameter, 10 μm thickness, 8-μm pores, Transwell; Costar) with the underside of the membranes coated with 10 μg/ml of collagen type I (Upstate Biotechnology) or BSA, as previously described (10, 21). In brief, COS7 cells, 40 h after transfection, or NMuMG cells stably expressing cDNAs, were trypsinized, washed, and suspended in fibroblast basal medium with 0.5% BSA at 1 × 106 cells/ml. Each 1 × 105 cells were then applied onto the upper migration chambers and allowed to migrate for 3 h. Cells migrated to the underside of the upper chamber were fixed with 3.7% paraformaldehyde in PBS, stained, and counted. COS7 cells positive for transfected cDNAs were detected by fluorescence from the EGFP-tag and counted by using a laser scanning microscope with a ×20 objective (model 510; Zeiss). Percent cell migration was calculated by dividing the number of migrated cells positive for transfection by the number of total applied cells positive for transfection. Each determination represents the average of triplicate of three independent experiments, and error bars represent the SEM.
Transcellular Invasion Assay of MM1 Cells.
In vitro transcellular invasion assay of MM1 cells was described previously (20, 22, 26). Briefly, after mesothelial cells from rat mesentery had reached confluency in a 35-mm dish, the culture medium was removed, and 2 × 105 MM1 cells were seeded onto the mesothelial cell monolayer in the modified minimum essential medium containing 25 μM lysophosphatidic acid (LPA), and allowed to migrate for 20 h. Penetrated single tumor cells and tumor cell colonies (invasion foci) were then counted under a phase contrast microscope (Olympus) in 16 different visual fields (each 0.59 mm2). In vitro invasion activities were quantitated as the percentage of infiltrated cells of the total cells initially applied, which was routinely about 10% with parental and vector control MM1 cells.
Growth Rate, Saturation Density, and Cell Size.
Cells (2 × 105) were plated onto 6-cm dishes (day 0) and grown for 5 days in the presence of serum. Cells were harvested by trypsinization, and viable cell counts were performed every 24 h. Cell sizes at day 1 were assessed by measuring the long and short axes on 20 cells fixed in 3.7% paraformaldehyde. Microscopic images at ×20 objective were then obtained with IX-70 (Olympus). Mouse epidermal growth factor (EGF) was purchased from Sigma.
BrdUrd Incorporation Assay.
Cells (1 × 105) were plated on 3.5-cm dishes and cultured for an additional 5 days. Cells were incubated with 10 μM BrdUrd (Sigma) for 3 h at various time points, and cells positive for BrdUrd were detected by using anti-BrdUrd antibody as described (27). Cells were also stained with 10 μg/ml 4′,6-diamidino-2′-phenylindole dihydrochloride (DAPI, Sigma) for 10 min to visualize the nucleus of all cell populations. Signals from BrdUrd and DAPI were detected by using laser scanning microscopy.
Data Analysis.
Statistical significance was examined by analysis of variance with post hoc multiple comparison by Fisher's multiple range test.
Immunoprecipitation and Immunoblotting Analysis.
Immunoprecipitations and immunoblotting analysis were described previously (28). Antibodies retained on the membranes were visualized by using an enzyme-linked chemiluminescence method (Amersham Pharmacia Biotech).
Results
Overexpression of Paxillin α, but Not Its Phosphorylation-Null Mutant, Reduced Cell Migratory Activities.
Tyrosine phosphorylation of paxillin α and p130Cas are the prominent events during integrin activation and cell migration (4). By using modified Boydem chambers, it has been shown that overexpressed p130Cas facilitates haptotactic cell migratory activity of COS7 cells toward several ECMs, including collagen, vitronectin, and fibronectin (10). No enhancement in migration has been seen with the phosphorylation-null mutation of p130Cas, suggesting that tyrosine phosphorylation of p130Cas plays an essential role. We then tested whether overexpression of paxillin α affected cell migration activity. As shown in Fig. 1A, overexpression of EGFP-paxillin α wild type (wt) reduced the haptotactic activity of COS7 cells toward collagen by >50%. Similar results were obtained on vitronectin and fibronectin (data not shown). On the other hand, such an effect was not observed with the phosphorylation-null mutant (4X mutant) of EGFP-paxillin α (Fig. 1A). The enhancing effect of p130Cas tagged with EGFP was confirmed (Fig. 1A), which was originally demonstrated by using a different tag (10). A previous report showed that the phosphorylation-null mutant of p130Cas (the ΔSD mutant) did not stimulate or inhibit the migration activity of COS7 cells on vitronectin (10). However, we observed that the ΔSD mutant significantly suppressed the migratory activity of COS7 cells toward collagen by >50% (Fig. 1A).
Figure 1.

Opposing effects of paxillin α and p130Cas on cell migratory activities through tyrosine phosphorylation. (A and B) Haptotactic transmigrating activities toward collagen type I by using modified Boyden chambers. Serum-starved COS7 cells (A) or serum-starved NMuMG cells (B) were allowed to migrate for 3 h on collagen-coated membrane after transfection with either the empty expression vector (vector) or the expression vector containing cDNA for EGFP-paxillin α wt (pax wt), its 4X mutant (pax 4X), EGFP-p130Cas wt (Cas wt), or its ΔSD mutant (Cas ΔSD). COS7 cells were transiently transfected by using FuGENE6 with an efficiency of 40–60%. NMuMG cells were stably transfected by retrovirus infection. Each bar represents the mean +/− SEM of triplicate of three independent experiments. (C) Transcellular invasion activities through mesothelial cell monolayer. LPA-stimulated MM1 cells cultured in the presence of 10% serum, stably expressing each cDNA as indicated, were seeded onto the mesothelial cell monolayer and allowed to migrate for 20 h without serum. Invasive activities of each transfectant were shown as relative activities to the vector control cells. Each bar represents the mean +/− SEM of triplicate of an independent experiments. Levels of exogenous proteins (open arrowheads) compared with endogenous proteins (closed arrowheads), as assessed by immunoblotting by using antipaxillin and anti-p130Cas, respectively, were also shown below (A–C).
Exogenous expression of EGFP-paxillin α wild type also reduced the haptotactic activity of serum-starved NMuMG cells toward collagen by about 40%, whereas EGFP-p130Cas wt increased it by about 300% as compared with the vector control (Fig. 1B). However, unlike the case of COS7 cells, expression of EGFP-paxillin α 4X increased the migratory activity by about 230%, whereas expression of EGFP-p130Cas ΔSD did not significantly change the activity (Fig. 1B). In these cells, each exogenous protein was stably expressed at a level comparable to each corresponding endogenous protein (Fig. 1B).
To examine further the negative effect of paxillin α expression on cell migratory activity, we also used an in vitro cell-monolayer invasion (transcellular migration) assay, in which the invasive capacity of MM1 hepatoma tumor cells is measured by counting cells penetrating a cultured mesothelial cell monolayer after tumor-cell seeding (20). In this system, serum-starved MM1 cells show high invasive activity when stimulated by LPA. As shown in Fig. 1C, expression of EGFP-paxillin α wt reduced the invasive activity of LPA-stimulated MM1 cells by >50%, whereas expression of the 4X mutant did not significantly affect the activity. The opposite effect was seen with expression of p130Cas. Although expression of EGFP-p130Cas wt was slightly inhibitory to the invasive activity, severe inhibition of the activity was observed when the ΔSD mutant was overexpressed. All of these transfected MM1 cells showed negligible invasive activity in the absence of LPA (data not shown).
In all of these experiments, protein expression was similar between EGFP-paxillin α wt and the 4X mutant and between EGFP-p130Cas wt and the ΔSD mutant (Fig. 1). Tyrosine phosphorylation of the wt exogenous proteins, but not the phosphorylation null-mutants, was also confirmed (data not shown).
Paxillin α and p130Cas Exert Opposing Effects on Cell Saturation Density Through Their Tyrosine Phosphorylation.
Integrin signaling is also involved in cell growth. We then measured the growth rates of these transfected NMuMG cells. As shown in Fig. 2, no significant differences in the growth rates among these cells were observed during their exponential growing phase (day 1 to day 3). However, there were large differences in cell densities at day 5, when cells had almost reached their plateau of saturation densities (Fig. 2; see also Fig. 3). Cells expressing EGFP-paxillin α wt stopped growing at densities almost half of those cells expressing the 4X mutant. Compared with the vector control, EGFP-paxillin α wt caused a decrease in the saturation density, whereas the phosphorylation-null mutant caused an increase. Apparently opposite phenomena were observed with EGFP-p130Cas. Expression of EGFP-p130Cas wt increased the cell saturation density, whereas the expression of the ΔSD mutant decreased it (Fig. 2B; also see Fig. 3). All-transfected cells formed contiguous flat monolayers but did not pile up or appeared transformed (Fig. 3).
Figure 2.
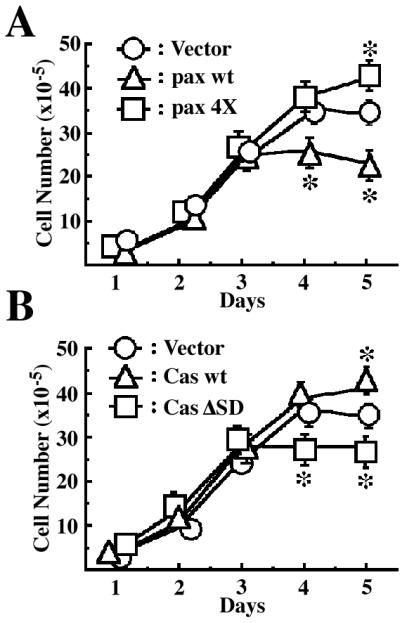
Effects of EGFP-paxillin α, EGFP-p130Cas, and their tyrosine phosphorylation null mutants on cell saturation densities. Each 2 × 105 cells were plated on 6-cm dishes and cultured for 5 days in the presence of serum. Cell numbers were measured every day by collecting cells and counting the viable cells. The same sets of NMuMG cells were used as in Fig. 1. Duplicate dishes were counted for each time point of the growth curves, and each determination represents the average of three independent experiments. Error bars represent the SEM. *, P < 0.05, as compared with the vector control cells.
Figure 3.

Properties of NMuMG cells expressing EGFP-paxillin α, EGFP-p130Cas, and their tyrosine phosphorylation null mutants. The same transfectants of NMuMG cells were used as in Fig. 2. (A) Phase-contrast microscopic images of same cells at days 1, 3, and 5, as in Fig. 3. Note that initial confluence was observed on day 3 with all of the transfectants. Piling up of cells was never observed even in prolonged culture more than 5 days. Bars = 100 μm. (B) Cell sizes measured at sparse cultures (day 1 as shown in Fig. 3). Mean lengths of long and short axis of cells are shown by hatched and solid bars, respectively. (C) BrdUrd incorporation of cells. Cells were incubated with 10 μM BrdUrd for 3 h at time, indicated, and then fixed. BrdUrd-positive cells were then visualized by using anti-BrdUrd antibody. Ratio of the BrdUrd-positive cells was calculated by visualizing nuclear DNA of all cell populations by using 4′,6-diamidino-2′-phenylindole dihydrochloride. Each determination represents the average of three independent experiments, and error bars represent the SEM.
The difference in the saturation densities among the different sets of transfected cells was not because of inherent changes in their cell shapes. Exogenous expression of neither paxillin α nor p130Cas cDNAs caused significant alterations in cell sizes or morphologies at sparse density (Fig. 3 A and B). A similar almost contiguous monolayer was seen at day 3 (Fig. 3A). However, as also shown in Fig. 2A, cells expressing EGFP-paxillin α wt seemed to almost stop growing after day 3, whereas cells expressing the 4X mutant continued to grow even after day 3, reaching higher density at day 5 (Fig. 3A). Incorporation of BrdUrd indicated that a larger fraction of cells expressing the 4X mutant continued to synthesize DNA during day 2 to day 4, as compared with cells expressing EGFP-paxillin α wt or the vector control (Fig. 3C). Again, opposing events were observed with EGFP-p130Cas wt and the ΔSD mutant in terms of the density-dependent regulation of cell growth and DNA synthesis, especially during day 3 to day 5 (Fig. 3 A and C).
Cell saturation density appears to be determined as an integration of several different extracellular stimuli, including those from growth factors. Addition of 10 nM of EGF to the vector control NMuMG cells further increased the saturation density by about 40%, as compared with that of the ordinary culture condition (10% serum) (Fig. 4): the addition of EGF to 100 nM had no further influence on growth or morphology, indicating that 5 × 106 cells in a 6-cm dish is the highest saturation density without being transformed. We found that the addition of EGF (10–100 nM) to NMuMG cells expressing the EGFP-paxillin α wt, cultured in the presence of 10% serum, increased the saturation density about 2-fold, to 5 × 106 cells in a 6-cm dish (Fig. 4; data not shown). The EGF-treated cells still formed a contiguous flat monolayer (data not shown). On the other hand, cells expressing EGFP-paxillin α 4X mutant or EGFP-p130Cas wt had already reached saturation of 5 × 106 cells when cultured in 10% serum, and no further increase in density was seen by the addition of 10 nM EGF (Fig. 4). However, it is noteworthy that the density of cells expressing EGFP-p130Cas ΔSD mutant could be increased only marginally by the addition of EGF (10–100 nM), and these cells never reached the higher density that other cells achieved (5 × 106 cells in a 6-cm dish) (Fig. 4).
Figure 4.
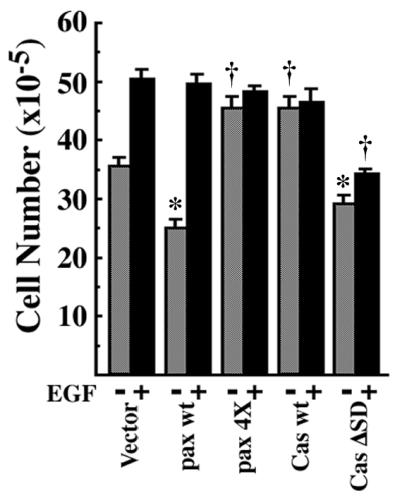
EGF increases cell saturation density of NMuMG cells expressing paxillin α. The same sets of NMuMG cells were used as in Fig. 2. Each 2 × 105 cells were plated on 6-cm dishes and cultured for 5 days in the presence of serum with (solid bar) or without (hatched bar) 10 nM EGF, and then viable cell numbers were counted. *, P < 0.05, †, P < 0.01 against values of the vector control.
Possible Different Signalings Downstream of Tyrosine Phosphorylation of Paxillin α and p130Cas.
Crk-II protein has been shown to be a major binding protein of tyrosine-phosphorylated p130Cas (29), which further increases p130Cas-mediated cell migration (10). Tyrosine-phosphorylated paxillin α has also been shown to bind to Crk-II and Crk-L in vivo (30, 31). We thus examined complex formation of p130Cas and paxillin α with c-Crk proteins in NMuMG cells. Epithelial–mesenchymal transdifferentiation is induced, and thus integrins are activated by TGF-β in NMuMG cells (19). As shown in Fig. 5, incubation with antibody against Crk-I/Crk-II precipitated tyrosine-phosphorylated proteins of 120–140 kDa in size, from both TGF-β-treated or untreated NMuMG cell lysates. Reblotting the membrane filter with other antibodies revealed that p130Cas, Abl, Cbl, and Fak were included in the immunoprecipitants. Increased binding was seen with p130Cas, Abl, and Cbl, but not with Fak, on TGF treatment. However, paxillin was not detected in the immunoprecipitants. Antibody against another Crk protein, Crk-L, also coprecipitated tyrosine-phosphorylated proteins of 120–140 kDa, but the identities of these proteins were unknown, and paxillin was again not included (Fig. 5). However, in an in vitro binding experiment, we confirmed strong and stable binding of the SH2 of Crk-I/Crk-II proteins to tyrosine-phosphorylated paxillin α in NMuMG cell lysates (data not shown). Therefore, these results indicate that tyrosine-phosphorylated paxillin α in NMuMG cells has a capacity to bind to Crk proteins, but unlike p130Cas, does not form a stable complex with Crk proteins in NMuMG cells.
Figure 5.
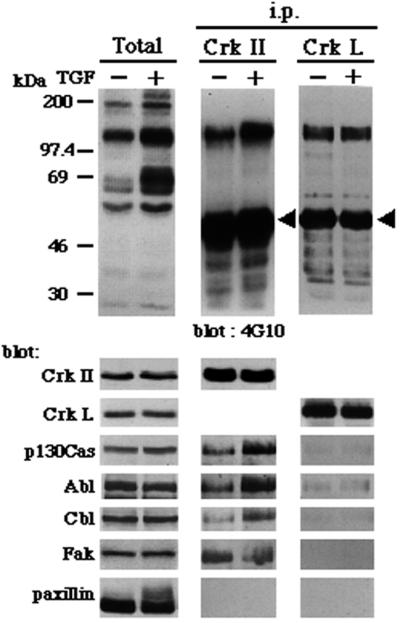
Paxillin does not stably associate with c-Crk proteins in NMuMG cells. NMuMG cells were treated or untreated with 2 ng/ml TGF-β for 3 days and then solubilized in 1% Nonidet P-40 buffer (27). Anti-Crk-I/Crk-II or anti-Crk-L immunoprecipitants were prepared from each 500 μg of 1% Nonidet P-40 cell lysates, separated by SDS/PAGE (8% gel), and subjected to sequential immunoblotting analysis by using antibodies as indicated. NMuMG cells expressed both Crk-I and Crk-II proteins (not shown). Total cell lysates (20 μg each) were also included (Total). Arrowheads indicate crossreaction of the secondary antibody with the heavy chain of anti-Crk antibodies used for immunoprecipitation.
Discussion
We showed in this paper that paxillin α and p130Cas potentially exert opposing effects on several integrin-mediated cellular events, possibly through tyrosine phosphorylation.
We showed that overexpression of EGFP-paxillin α reduces cell migratory activities, whereas overexpression of EGFP-p130Cas enhances them. Such phenomena were observed in haptotactic cell migration activities of NMuMG cells and COS7 cells toward ECMs, in the absence of serum. Tyrosine phosphorylation-null mutations of each protein abolished these effects or exhibited dominant-negative effects in several assays. For example, expression of the 4X mutant of paxillin α enhanced the cell migration activity of NMuMG cells, whereas expression of the ΔSD mutant of p130Cas reduced the cell migration activity of COS7 cells. Overexpression of EGFP-paxillin α also reduced the in vitro transcellular invasion activity of MM1 cells penetrating mesothelial cell monolayers in the presence of LPA, whereas such effect was not seen with overexpression of the phosphorylation null-mutation of paxillin α. Unlike the haptotactic cell migration assays described above, on the other hand, EGFP-p130Cas did not show enhanced transcellular invasive activity of MM1 cells. However, severe inhibition was observed when the phosphorylation null-mutant p130Cas was expressed. Other investigators have also shown in U-87MG glioblastoma cells that p130Cas does not act to enhance cell migratory activity, but the ΔSD mutant reduces it (11). We thus concluded that tyrosine phosphorylation of p130Cas and paxillin α appears to exert opposing effects on cell migratory activities. EGFP-tagged paxillin α and p130Cas, as well as the 4X mutant and the ΔSD mutant, showed similar subcellular localization to each corresponding endogenous protein in NMuMG cells (unpublished data).
Cell migratory signaling after the tyrosine phosphorylation of p130Cas has been relatively well studied, as already described in this paper. The current model is that Fak-mediated tyrosine phosphorylation of p130Cas leads to Crk binding and possibly to Rac activation (9, 10). On the other hand, whereas a recent publication suggests that Crk II protein may be a downstream effector of paxillin in Nara Bladder Tumor-II bladder tumor cells (32), only a limited number of studies have been available with regards to cell migration signaling downstream of paxillin tyrosine phosphorylation. We showed that paxillin α and p130Cas exert opposing effects on cell migratory activities. Therefore, analysis of the paxillin α-mediated intracellular signaling, particularly in comparison with that of p130Cas within the same cellular contexts, would greatly facilitate our understanding of cell migratory signaling.
Our results also indicate that tyrosine phosphorylation of paxillin α and p130Cas affect cell saturation densities of NMuMG cells in opposing ways. Expression of EGFP-paxillin α reduced the saturation density, whereas EGFP-p130Cas increased it. Moreover, tyrosine phosphorylation-null mutants acted as dominant negatives in each case: the 4X mutant of paxillin α increased the saturation density, whereas the ΔSD mutant of p130Cas decreased it, as compared with the vector control. Our results thus suggest that tyrosine phosphorylation of both paxillin α and p130Cas is also involved in the contact inhibition of growth. Contact inhibition of growth plays an essential role in contiguous flat monolayer formation, and loss of contact inhibition has been well documented to be closely related to oncogenic cell transformation, including that by v-Src and v-Crk, where aberrant tyrosine phosphorylation of paxillin and p130Cas has taken place (33–35). Contact inhibition of growth may be determined as a consequence of the coordination of several different extracellular stimuli, including those from growth factors, from integrin-mediated adhesion, and possibly from cell–cell contacts. Continuation of integrin-mediated cell migration may play a crucial role in efficiently forming cell–cell contacts sufficiently for the occurrence of contact inhibition of growth. It has been shown that cell migration and growth use many of the same intracellular signaling pathways, including activation of Rho family GTPases and MAP kinase (extracellular signal-regulated protein kinase 1/2) cascade (36–40). Moreover, growth factor stimulation such as by EGF or platelet-derived growth factor has been shown to affect tyrosine phosphorylation of p130Cas and paxillin (41–44), as well as cell migration (45, 46). We showed that addition of EGF could increase the saturation density of NMuMG cells expressing EGFP-paxillin α, thus indicating that paxillin α signaling pathways intercommunicate with those from the EGF receptor to determine the threshold of contact inhibition of growth.
Both p130Cas and paxillin α contain multiple tyrosine phosphorylation sites bearing a Tyr-X-X-Pro motif, a predicted strong binding sequence to several SH2 including those of Crk and Nck (6). Crk and Nck have been identified as direct downstream effectors of tyrosine phosphorylation of p130Cas (8, 29, 47). Crk SH2 has been shown also to bind to tyrosine-phosphorylated paxillin (48, 49). Butler et al. (31) have shown that paxillin is coprecipitated with CrkII and CrkL in lysates prepared from rat uterus after the rats were treated with insulin-like growth factor-I. Salgia et al. (30) have also demonstrated that CrkL binds to paxillin in vivo through the SH2. We have confirmed in vitro that the SH2 of Crk-II indeed stably bind to paxillin tyrosine-phosphorylated at Tyr-31 and Tyr-118 residues (unpublished results), both of which are highly phosphorylated during active cell migration of NMuMG cells (4). However, we could obtain no firm evidence showing stable intracellular binding of Crk-I/Crk-II or CrkL toward phosphorylated paxillin α in NMuMG cells, although we have used antibodies from the same commercial sources as previously described (30, 31). No significant association of tyrosine-phosphorylated paxillin with Crk-I/Crk-II has been reported with other types of cells (30, 50). We thus conclude that paxillin α does not form a stable complex with CrkI/Crk II or CrkL proteins in NMuMG cells. We also did not obtain evidence for the binding of paxillin α to Nck in NMuMG cells (unpublished results). However, the possibility still remains that paxillin α can interact with these proteins in vivo with biochemical kinetics quite different from those of p130Cas interaction; for example, tyrosine-phosphorylated paxillin α may interact with Crk proteins through very high association and dissociation rates in NMuMG cells. Further careful examination should be conducted to determine the bona fide downstream effectors of paxillin tyrosine phosphorylation in vivo. Our future efforts will thus focus on identifying downstream effectors of paxillin tyrosine phosphorylation, and delineating and dissecting the intracellular signaling, that regulate cell migratory activities as well as contact inhibition of growth by using paxillin and p130Cas as molecular tools.
Acknowledgments
We are grateful to Hisamaru Hirai (Tokyo University, Tokyo) for p130Cas cDNAs and anti-p130Cas antibody, to Warren Pear and David Baltimore (California Institute of Technology, Pasadena, CA) for BOSC23 cells, and to Hartmut Land (Imperial Cancer Research Fund, London) for pBabe vector. We also thank Mihoko Sato and Manami Hiraishi for their technical assistance, Mayumi Yoneda for secretarial work, and Kathy Barker for critical reading of the manuscript. This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports, and Culture of Japan; by grants from the Takeda Medical Foundation, the Mitsubishi Foundation, the Ciba–Geigy Foundation (Japan) for the Promotion of Science, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Novartis Foundation for the Promotion of Science.
Abbreviations
- p130Cas
Crk-associated substrate
- ECM
extracellular matrices
- NMuMG cell
normal murine mammary gland cell
- Fak
focal adhesion kinase
- SH2
src homology domain 2
- EGFP
enhanced green fluorescent protein
- 4X mutant
phosphorylation-null mutant of paxillin
- ΔSD mutant
phosphorylation-null mutant of p130Cas
- LPA
lysophosphatidic acid
- EGF
epidermal growth factor
- wt
wild type
- TGF-β
transforming growth factor β
References
- 1.Hynes R O. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 2.Lauffenburger D A, Horwitz A, F. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 3.Sheetz M P, Felsenfeld D P, Galbraith C G. Trends Cell Biol. 1998;8:51–54. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura, K., Yano, H., Uchida, H., Hashimoto, S., Schaefer, E. & Sabe, H. J. (2000) J. Biol. Chem., in press. [DOI] [PubMed]
- 5.Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. EMBO J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Songyang Z, Shoelson S E, Chaudhuri M, Gish G, Pawson T, Haser W G, King F, Roberts T, Ratnofsky S, Lechleider R J, et al. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 7.Vuori K, Hirai H, Aizawa S, Ruoslahti E. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlaepfer D D, Broome M A, Hunter T. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cary L A, Han D C, Polte T R, Hanks S K, Guan J L. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klemke R L, Leng J, Molander R, Brooks P C, Vuori K, Cheresh D A. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu J, Tamura M, Pankov R, Danen E H J, Takino T, Matsumoto K, Yamada K M. J Cell Biol. 1999;146:389–403. doi: 10.1083/jcb.146.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark E A, Brugge J S. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 13.Turner C E. Int J Biochem Cell Biol. 1998;30:955–959. doi: 10.1016/s1357-2725(98)00062-4. [DOI] [PubMed] [Google Scholar]
- 14.Burridge K, Turner C E, Romer L H. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richardson A, Malik R K, Hildebrand J D, Parsons J T. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham I L, Anderson D C, Holers V M, Brown E J. J Cell Biol. 1994;127:1139–1147. doi: 10.1083/jcb.127.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellis S L, Miller J T, Turner C E. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- 18.Thomas S M, Soriano P, Imamoto A. Nature (London) 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- 19.Miettinen P J, Ebner R, Lopez A R, Derynck R. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akedo H, Shinkai K, Mukai M, Mori Y, Tateishi R, Tanaka K, Yamamoto R, Morishita T. Cancer Res. 1986;46:2416–2422. [PubMed] [Google Scholar]
- 21.Kondo A, Hashimoto S, Yano H, Nagayama K, Mazaki Y, Sabe H. Mol Biol Cell. 2000;11:1315–1327. doi: 10.1091/mbc.11.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukai M, Imamura F, Ayaki M, Shinkai K, Iwasaki T, Murakami-Murofushi K, Murofushi H, Kobayashi S, Yamamoto T, Nakamura H, Akedo H. Int J Cancer. 1999;81:918–922. doi: 10.1002/(sici)1097-0215(19990611)81:6<918::aid-ijc13>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Mazaki Y, Uchida H, Hino O, Hashimoto S, Sabe H. J Biol Chem. 1998;273:22435–22441. doi: 10.1074/jbc.273.35.22435. [DOI] [PubMed] [Google Scholar]
- 24.Mayer B J, Hirai H, Sakai R. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 25.Pear W S, Nolan G P, Scott M L, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshioka K, Matsumura F, Akedo H, Itoh K. J Biol Chem. 1998;273:5146–5154. doi: 10.1074/jbc.273.9.5146. [DOI] [PubMed] [Google Scholar]
- 27.Vinals F, Pouyssegur J. Mol Cell Biol. 1999;19:2763–2772. doi: 10.1128/mcb.19.4.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Proc Natl Acad Sci USA. 1994;91:3984–3988. doi: 10.1073/pnas.91.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamoto T, Sakai R, Ozawa K, Yazaki Y, Hirai H. J Biol Chem. 1996;271:8959–8965. doi: 10.1074/jbc.271.15.8959. [DOI] [PubMed] [Google Scholar]
- 30.Salgia R, Uemura N, Okuda K, Li J L, Pisick E, Sattler M, Jong R, Druker B, Heisterkamp N, Chen L B, Groffen J, Griffin J D. J Biol Chem. 1995;270:29145–29150. doi: 10.1074/jbc.270.49.29145. [DOI] [PubMed] [Google Scholar]
- 31.Butler A A, Blakesley V A, Koval A, Jong R, Groffen J, LeRoith D. J Biol Chem. 1997;272:27660–27664. doi: 10.1074/jbc.272.44.27660. [DOI] [PubMed] [Google Scholar]
- 32.Petit V, Boyer B, Lentz D, Turner C E, Thiery J P, Valles A M. J Cell Biol. 2000;148:957–969. doi: 10.1083/jcb.148.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer B J, Hamaguchi M, Hanafusa H. Nature (London) 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 34.Glenney J R, Zokas L. J Cell Biol. 1989;108:2401–2408. doi: 10.1083/jcb.108.6.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner C E, Glenney J R, Burridge K. J Cell Biol. 1990;111:1059–1068. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pages G, Lenormand P, L'Allemain G, Chambard J C, Meloche S, Pouyssegur J. Proc Natl Acad Sci USA. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsen M F, Ashworth A, Hall A. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 38.Anad-Apte B, Zetter B R, Viswanathan A, Qiu R G, Chen J, Ruggieri R, Symons M. J Biol Chem. 1997;272:30688–30692. doi: 10.1074/jbc.272.49.30688. [DOI] [PubMed] [Google Scholar]
- 39.Klemke R L, Cai S, Giannini A L, Gallagher P J, Lanerolle P, Cheresh D A. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins L R, Ricketts W A, Yeh L, Cheresh D. J Cell Biol. 1999;147:1561–1568. doi: 10.1083/jcb.147.7.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ojaniemi M, Vuori K. J Biol Chem. 1997;272:25993–25998. doi: 10.1074/jbc.272.41.25993. [DOI] [PubMed] [Google Scholar]
- 42.Tapia J A, Camello C, Jensen R T, Garcia L J. Biochem Biophys Acta. 1999;1448:486–499. doi: 10.1016/s0167-4889(98)00157-8. [DOI] [PubMed] [Google Scholar]
- 43.Casamassima A, Rozengurt E. J Biol Chem. 1997;272:9363–9370. doi: 10.1074/jbc.272.14.9363. [DOI] [PubMed] [Google Scholar]
- 44.Rankin S, Rozengurt E. J Biol Chem. 1994;269:704–710. [PubMed] [Google Scholar]
- 45.Wells A. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 46.Herdin C-H, Westermark B. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 47.Hamasaki K, Mimura T, Morino N, Furuya H, Nakamoto T, Aizawa S, Morimoto C, Yazaki Y, Hirai H, Nojima Y. Biochem Biophys Res Commun. 1996;222:338–343. doi: 10.1006/bbrc.1996.0745. [DOI] [PubMed] [Google Scholar]
- 48.Birge R B, Fajardo J E, Reichman C, Shoelson S E, Songyang Z, Cantley L C, Hanafusa H. Mol Cell Biol. 1993;13:4648–4656. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabe H, Shoelson S E, Hanafusa H. J Biol Chem. 1995;270:31219–31224. doi: 10.1074/jbc.270.52.31219. [DOI] [PubMed] [Google Scholar]
- 50.Nakashima N, Rose D W, Xiao S, Egawa K, Martin S S, Haruta T, Saltiel A R, Olefsky J M. J Biol Chem. 1999;274:3001–3008. doi: 10.1074/jbc.274.5.3001. [DOI] [PubMed] [Google Scholar]