

Abstract
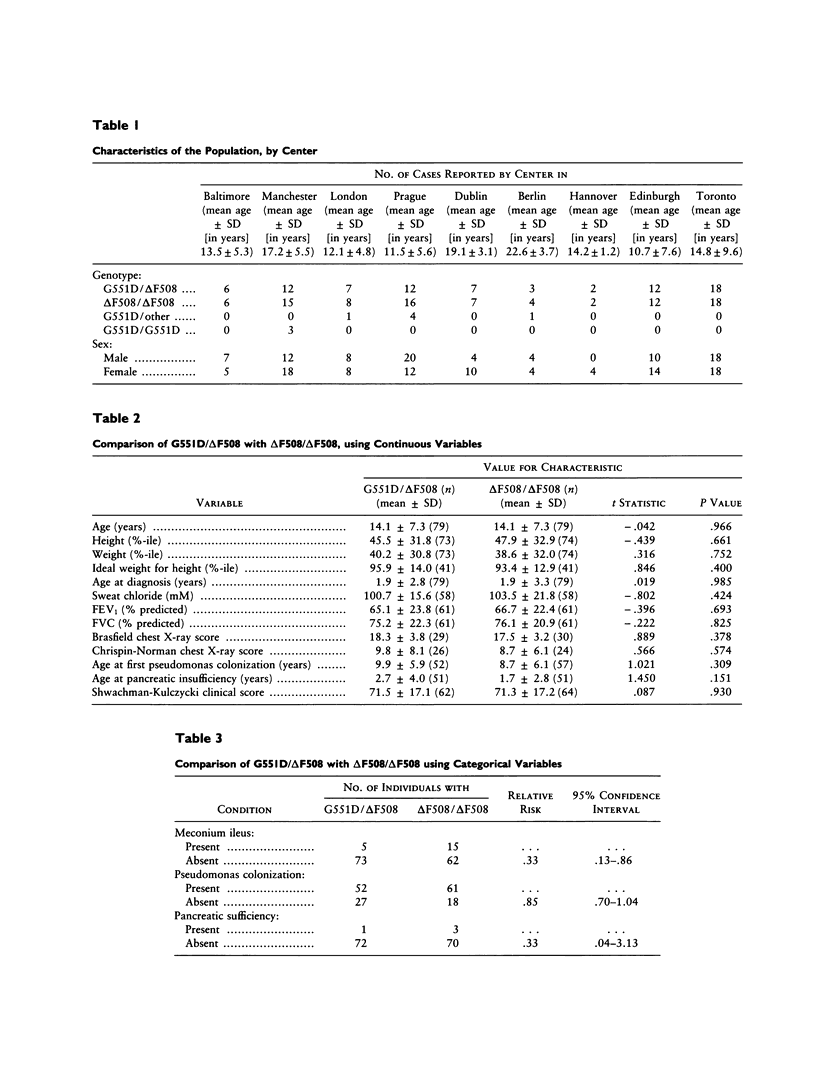
The glycine-to-aspartic acid missense mutation at codon 551 (G551D), which is within the first nucleotide-binding fold of the cystic fibrosis transmembrane conductance regulator (CFTR), is the third most common cystic fibrosis (CF) mutation, with a worldwide frequency of 3.1% among CF chromosomes. Regions with a high frequency correspond to areas with large populations of Celtic descent. To determine whether G551D confers a different phenotype than does ΔF508, the most common CF mutation, we studied 79 compound heterozygotes for G551D/ΔF508, from nine centers in Europe and North America. Each subject was matched, by age and sex, with a ΔF508 homozygote from the same center. A retrospective cohort analysis was performed on the following outcome parameters: age at diagnosis, sweat chloride, meconium ileus at birth, height, weight, weight for height, FVC, FEV1, chest X-ray score, pseudomonas colonization, pancreatic sufficiency, and Shwachman clinical score. There was less meconium ileus among the G551D/ΔF508 compound heterozygotes (relative risk 0.33; 95% confidence interval .13–.86), as well as a trend toward later age at diagnosis of pancreatic insufficiency. No statistically significant difference was found between the groups for any other parameter. These results suggest that the CF genotype can be a predictor of pancreatic and intestinal phenotype. Prenatal counseling for the two genotype groups should differ only with respect to probability of meconium ileus. Clinical outcome (after survival of meconium ileus) for G551D/ΔF508 compound heterozygotes and ΔF508 homozygotes is indistinguishable; therefore, prognostic counseling should not differ.
Full text
PDF




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Brasfield D., Hicks G., Soong S., Tiller R. E. The chest roentgenogram in cystic fibrosis: a new scoring system. Pediatrics. 1979 Jan;63(1):24–29. [PubMed] [Google Scholar]
- Campbell P. W., 3rd, Phillips J. A., 3rd, Krishnamani M. R., Maness K. J., Hazinski T. A. Cystic fibrosis: relationship between clinical status and F508 deletion. J Pediatr. 1991 Feb;118(2):239–241. doi: 10.1016/s0022-3476(05)80490-1. [DOI] [PubMed] [Google Scholar]
- Chrispin A. R., Norman A. P. The systematic evaluation of the chest radiograph in cystic fibrosis. Pediatr Radiol. 1974;2(2):101–105. doi: 10.1007/BF01314939. [DOI] [PubMed] [Google Scholar]
- Cutting G. R., Kasch L. M., Rosenstein B. J., Zielenski J., Tsui L. C., Antonarakis S. E., Kazazian H. H., Jr A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein. Nature. 1990 Jul 26;346(6282):366–369. doi: 10.1038/346366a0. [DOI] [PubMed] [Google Scholar]
- Johansen H. K., Nir M., Høiby N., Koch C., Schwartz M. Severity of cystic fibrosis in patients homozygous and heterozygous for delta F508 mutation. Lancet. 1991 Mar 16;337(8742):631–634. doi: 10.1016/0140-6736(91)92449-c. [DOI] [PubMed] [Google Scholar]
- Kerem B. S., Zielenski J., Markiewicz D., Bozon D., Gazit E., Yahav J., Kennedy D., Riordan J. R., Collins F. S., Rommens J. M. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc Natl Acad Sci U S A. 1990 Nov;87(21):8447–8451. doi: 10.1073/pnas.87.21.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989 Sep 8;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Kerem E., Corey M., Kerem B. S., Rommens J., Markiewicz D., Levison H., Tsui L. C., Durie P. The relation between genotype and phenotype in cystic fibrosis--analysis of the most common mutation (delta F508). N Engl J Med. 1990 Nov 29;323(22):1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- Kerem E., Corey M., Kerem B., Durie P., Tsui L. C., Levison H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J Pediatr. 1989 May;114(5):767–773. doi: 10.1016/s0022-3476(89)80134-9. [DOI] [PubMed] [Google Scholar]
- Olsen M. M., Gauderer M. W., Girz M. K., Izant R. J., Jr Surgery in patients with cystic fibrosis. J Pediatr Surg. 1987 Jul;22(7):613–618. doi: 10.1016/s0022-3468(87)80111-2. [DOI] [PubMed] [Google Scholar]
- SHWACHMAN H., KULCZYCKI L. L. Long-term study of one hundred five patients with cystic fibrosis; studies made over a five- to fourteen-year period. AMA J Dis Child. 1958 Jul;96(1):6–15. doi: 10.1001/archpedi.1958.02060060008002. [DOI] [PubMed] [Google Scholar]
- Santis G., Osborne L., Knight R. A., Hodson M. E. Independent genetic determinants of pancreatic and pulmonary status in cystic fibrosis. Lancet. 1990 Nov 3;336(8723):1081–1084. doi: 10.1016/0140-6736(90)92566-z. [DOI] [PubMed] [Google Scholar]