

Abstract
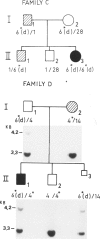
Hyperphenylalaninemia (HPA) results from defective hydroxylation of phenylalanine in the liver, in most cases because of defective phenylalanine hydroxylase. HPA is highly variable, ranging from moderate elevation of plasma phenylalanine with no clinical consequences to a severe disease, classical phenylketonuria (PKU). Non-PKU HPA was found in excess of PKU in Israel, while the opposite is true in Europe. To study the genetic basis of non-PKU HPA, we performed haplotype analysis at the phenylalanine hydroxylase locus in 27 families with non-PKU HPA. All individuals with this condition were compound heterozygotes. In six of these families, in which both PKU and non-PKU HPA were segregating, haplotype analysis showed that non-PKU HPA resulted from compound heterozygosity for a PKU mutation and a second mutation, with milder effect, which is probably expressed only when it interacts with the severe mutation. The involvement of PKU mutations in non-PKU HPA was further demonstrated in Jewish Yemenite families with non-PKU HPA, in which the individuals with this condition were carriers of the single PKU allele which exists in this community. In addition, two previously known PKU point mutations (R261Q and R408W) were found in individuals with non-PKU HPA. These mutations are associated, in our population, with the same haplotypes as those with which it is associated in Europe. Based on the above-mentioned genetic model for non-PKU HPA, successful prenatal diagnosis of this condition was performed in one family.(ABSTRACT TRUNCATED AT 250 WORDS)
Full text
PDF







Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson J. A., Fisch R., Miller E., Doeden D. Atypical phenylketonuric heterozygote. Deficiency in phenylalanine hydroxylase and transaminase activity. J Pediatr. 1966 Mar;68(3):351–360. doi: 10.1016/s0022-3476(66)80237-8. [DOI] [PubMed] [Google Scholar]
- Avigad S., Cohen B. E., Bauer S., Schwartz G., Frydman M., Woo S. L., Niny Y., Shiloh Y. A single origin of phenylketonuria in Yemenite Jews. Nature. 1990 Mar 8;344(6262):168–170. doi: 10.1038/344168a0. [DOI] [PubMed] [Google Scholar]
- Bartholomé K., Lutz P., Bickel H. Determination of phenylalanine hydroxylase activity in patients with phenylketonuria and hyperphenylalaninemia. Pediatr Res. 1975 Dec;9(12):899–903. doi: 10.1203/00006450-197512000-00006. [DOI] [PubMed] [Google Scholar]
- Bartholomé K., Olek K., Trefz F. Compound heterozygotes in hyperphenylalaninaemia. Hum Genet. 1984;65(4):405–406. doi: 10.1007/BF00291569. [DOI] [PubMed] [Google Scholar]
- Berman J. L., Cunningham G. C., Day R. W., Ford R., Hsia D. Y. Causes for high phenylalanine with normal tyrosine in newborn screening programs. Am J Dis Child. 1969 Jan;117(1):54–65. doi: 10.1001/archpedi.1969.02100030056006. [DOI] [PubMed] [Google Scholar]
- Blaskovics M. E., Schaeffler G. E., Hack S. Phenylalaninaemia. Differential diagnosis. Arch Dis Child. 1974 Nov;49(11):835–843. doi: 10.1136/adc.49.11.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R., Lidsky A. S., Daiger S. P., Güttler F., Sullivan S., Dilella A. G., Woo S. L. Polymorphic DNA haplotypes at the human phenylalanine hydroxylase locus and their relationship with phenylketonuria. Hum Genet. 1987 May;76(1):40–46. doi: 10.1007/BF00283048. [DOI] [PubMed] [Google Scholar]
- Cohen B. E., Szeinberg A., Levine Y., Peled I., Pollack S., Crispin M., Normand M. Phenylketonuria (PKU) in Israel. Monogr Hum Genet. 1978;9:95–101. doi: 10.1159/000401617. [DOI] [PubMed] [Google Scholar]
- Coutts N. A., Fyfe W. M. Classical and mild phenylketonuria in a family. Arch Dis Child. 1971 Aug;46(248):550–552. doi: 10.1136/adc.46.248.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firon N., Eyal N., Kolodny E. H., Horowitz M. Genotype assignment in Gaucher disease by selective amplification of the active glucocerebrosidase gene. Am J Hum Genet. 1990 Mar;46(3):527–532. [PMC free article] [PubMed] [Google Scholar]
- GUTHRIE R., SUSI A. A SIMPLE PHENYLALANINE METHOD FOR DETECTING PHENYLKETONURIA IN LARGE POPULATIONS OF NEWBORN INFANTS. Pediatrics. 1963 Sep;32:338–343. [PubMed] [Google Scholar]
- Hsia D. Y., O'Flynn M. E., Berman J. L. Atypical phenylketonuria with borderline or normal intelligence. Am J Dis Child. 1968 Aug;116(2):143–157. doi: 10.1001/archpedi.1968.02100020145005. [DOI] [PubMed] [Google Scholar]
- John S. W., Rozen R., Scriver C. R., Laframboise R., Laberge C. Recurrent mutation, gene conversion, or recombination at the human phenylalanine hydroxylase locus: evidence in French-Canadians and a catalog of mutations. Am J Hum Genet. 1990 May;46(5):970–974. [PMC free article] [PubMed] [Google Scholar]
- Kang E. S., Kaufman S., Gerald P. S. Clinical and biochemical observations of patients with atypical phenylketonuria. Pediatrics. 1970 Jan;45(1):83–92. [PubMed] [Google Scholar]
- Kaufman S., Max E. E., Kang E. S. Phenylalanine hydroxylase activity in liver biopsies from hyperphenylalaninemia heterozygotes: deviation from proportionality with gene dosage. Pediatr Res. 1975 Aug;9(8):632–634. doi: 10.1203/00006450-197508000-00004. [DOI] [PubMed] [Google Scholar]
- Ledley F. D., Levy H. L., Woo S. L. Molecular analysis of the inheritance of phenylketonuria and mild hyperphenylalaninemia in families with both disorders. N Engl J Med. 1986 May 15;314(20):1276–1280. doi: 10.1056/NEJM198605153142002. [DOI] [PubMed] [Google Scholar]
- Leeming R. J., Barford P. A., Blair J. A., Smith I. Blood spots on Guthrie cards can be used for inherited tetrahydrobiopterin deficiency screening in hyperphenylalaninaemic infants. Arch Dis Child. 1984 Jan;59(1):58–61. doi: 10.1136/adc.59.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter-Konecki U., Schlotter M., Konecki D. S., Labeit S., Woo S. L., Trefz F. K. Linkage disequilibrium between mutation and RFLP haplotype at the phenylalanine hydroxylase locus in the German population. Hum Genet. 1988 Apr;78(4):347–352. doi: 10.1007/BF00291733. [DOI] [PubMed] [Google Scholar]
- Navon R., Kolodny E. H., Mitsumoto H., Thomas G. H., Proia R. L. Ashkenazi-Jewish and non-Jewish adult GM2 gangliosidosis patients share a common genetic defect. Am J Hum Genet. 1990 Apr;46(4):817–821. [PMC free article] [PubMed] [Google Scholar]
- Niederwieser A., Curtius H. C., Wang M., Leupold D. Atypical phenylketonuria with defective biopterin metabolism. Monotherapy with tetrahydrobiopterin or sepiapterin, screening and study of biosynthesis in man. Eur J Pediatr. 1982 Mar;138(2):110–112. doi: 10.1007/BF00441135. [DOI] [PubMed] [Google Scholar]
- Okano Y., Wang T., Eisensmith R. C., Güttler F., Woo S. L. Recurrent mutation in the human phenylalanine hydroxylase gene. Am J Hum Genet. 1990 May;46(5):919–924. [PMC free article] [PubMed] [Google Scholar]
- Rey F., Berthelon M., Caillaud C., Lyonnet S., Abadie V., Blandin-Savoja F., Feingold J., Saudubray J. M., Frézal J., Munnich A. Clinical and molecular heterogeneity of phenylalanine hydroxylase deficiencies in France. Am J Hum Genet. 1988 Dec;43(6):914–921. [PMC free article] [PubMed] [Google Scholar]
- Sakai K., Kanda N., Shiloh Y., Donlon T., Schreck R., Shipley J., Dryja T., Chaum E., Chaganti R. S., Latt S. Molecular and cytologic analysis of DNA amplification in retinoblastoma. Cancer Genet Cytogenet. 1985 Jun;17(2):95–112. doi: 10.1016/0165-4608(85)90020-2. [DOI] [PubMed] [Google Scholar]
- Sullivan S. E., Moore S. D., Connor J. M., King M., Cockburn F., Steinmann B., Gitzelmann R., Daiger S. P., Woo S. L. Haplotype distribution of the human phenylalanine hydroxylase locus in Scotland and Switzerland. Am J Hum Genet. 1989 May;44(5):652–659. [PMC free article] [PubMed] [Google Scholar]
- Trefz F. K., Bartholomé K., Bickel H., Lutz P., Schmidt H., Seyberth H. W. In vivo residual activities of the phenylalanine hydroxylating system in phenylketonuria and variants. J Inherit Metab Dis. 1981;4(2):101–102. doi: 10.1007/BF02263611. [DOI] [PubMed] [Google Scholar]
- WONG P. W., O'FLYNN M. E., INOUYE T. MICROMETHODS FOR MEASURING PHENYLALANINE AND TYROSINE IN SERUM. Clin Chem. 1964 Dec;10:1098–1104. [PubMed] [Google Scholar]
- Wang T., Okano Y., Eisensmith R. C., Fekete G., Schuler D., Berencsi G., Nasz I., Woo S. L. Molecular genetics of PKU in eastern Europe: a nonsense mutation associated with haplotype 4 of the phenylalanine hydroxylase gene. Somat Cell Mol Genet. 1990 Jan;16(1):85–90. doi: 10.1007/BF01650483. [DOI] [PubMed] [Google Scholar]
- Woo S. L. Collation of RFLP haplotypes at the human phenylalanine hydroxylase (PAH) locus. Am J Hum Genet. 1988 Nov;43(5):781–783. [PMC free article] [PubMed] [Google Scholar]
- Woolf L. I., Cranston W. I., Goodwin B. L. Genetics of phenylketonuria. Heterozygosity for phenylketonuria. Nature. 1967 Mar 4;213(5079):882–883. doi: 10.1038/213882a0. [DOI] [PubMed] [Google Scholar]
- Woolf L. I., Goodwin B. L., Cranston W. I., Wade D. N., Woolf F., Hudson F. P., McBean M. S. A third allele at the phenylalanine-hydroxylase locus in mild phenylketonuria (hyperphenylalaninaemia). Lancet. 1968 Jan 20;1(7534):114–117. doi: 10.1016/s0140-6736(68)92722-0. [DOI] [PubMed] [Google Scholar]