

Abstract
Based on the DNA sequence of the symbiotic plasmid of Rhizobium strain NGR234, we predicted potential rearrangements generated by homologous recombination. All predicted rearrangements were identified experimentally by using a PCR-based methodology. Thus, the predicted and the actual dynamic maps of the replicon coincide. By using an approach that does not involve the introduction of exogenous genetic elements, derivative populations that are pure for specific rearrangements were obtained. We propose that knowledge of the DNA sequence of a genome offers the possibility of designing pathways of sequential rearrangements leading to alternative genomic structures. An experimental strategy to isolate bacterial populations containing the desired structures is discussed.
Reiterated DNA sequences in a genome are potential sites for homologous recombination which results in the generation of rearrangements (1, 2). Recombination between direct-repeated sequences leads to either amplification (amp) or deletion (del) of DNA, whereas recombination between inverted sequences results in inversion. Recombination between repeated sequences present in different replicons may lead to their cointegration. Some of these rearrangements may have important biological consequences. In particular, DNA amp has been associated with adaptative responses in different bacteria (3, 4).
The past 5 years have witnessed a veritable explosion in the number of completely sequenced genomes, thus heralding a new era in genomic research (5). Besides the immediate knowledge and extrapolations that can be gained from that information, powerful approaches for whole-genome analysis are being generated (6). An interesting extension of this information relates to genome dynamics. If the position and orientation of the reiterated DNA families of a genome or a region of a genome are known, the different types of potential rearrangements produced by homologous recombination may be predicted. In this work, we studied the symbiotic plasmid (pSym) of Rhizobium strain NGR234 as a model system to test this view.
Bacteria of the genus Rhizobium interact with the roots of leguminous plants and establish nitrogen fixing symbioses. Strain NGR234 is of particular biological interest because it possesses the broadest host range of any strain so far analyzed. It is able to nodulate legumes from more than 110 genera as well as the nonlegume Parasponia andersonii (7). The genome of this strain consists of three replicons: the chromosome, the pSym or pNGR234a, and a megaplasmid or pNGR234b (8). The complete nucleotide sequence of pNGR234a has been reported (9). This replicon is a circular structure of 536,165 bp and contains most of the genes for nodulation and nitrogen fixation.
Based on the DNA sequence of pNGR234a, we predicted and experimentally confirmed the occurrence of different genomic rearrangements originated by homologous recombination. Subpopulations of the wild-type Rhizobium strain NGR234, pure for specific rearrangements, were isolated and characterized.
Materials and Methods
Culture Conditions.
Rhizobium strain NGR234 and its derivatives were grown at 28°C in PY medium (0.5% peptone/0.3% yeast extract/10 mM CaCl2) containing 50 μg/ml rifampicin.
DNA Manipulations.
Total DNA was purified by using a DNA/RNA isolation kit (USB; Amersham Pharmacia). Plasmid profiles of Rhizobium were obtained as described by Hynes and McGregor (10). Standard DNA manipulations such as restriction, agarose gel electrophoresis, and filter blot hybridization were performed as previously described (11). Probes were radiolabeled with 32P with a random-priming DNA labeling kit (rediprime; Amersham Pharmacia).
Oligonucleotide Primers and PCR Assays.
All of the primers used were 30-mer and were commercially synthesized by Biosynthesis (Lewinsville, TX). The exact position of the 5′ start base of each primer is indicated according to the reported sequence of pNGR234a (9): NGRIS4a forward primer (FP), 18,270; NGRIS4a reverse primer (RP), 23,289; NGRIS5a FP, 186,835; NGRIS5a RP, 193,303; NGRIS3b FP, 216,592; NGRIS3b RP, 220,719; NGRIS5b FP, 280,018; NGRIS5b RP, 283,273; NGRIS2a FP, 319,737; NGRIS2a RP, 323,376; NGRIS4b FP, 319,996; NGRIS4b RP, 330,276; NGRIS2b FP, 376,892; NGRIS2b RP, 380,543; NGRIS5c FP, 380,534; NGRIS5c RP, 386,277; NGRIS3c FP, 399,731; NGRIS3c RP, 403,870; NGRnifHDK1 FP, 451,460; NGRnifHDK1 RP, 456,186; NGRnifHDK2 FP, 483,685; and NGRnifHDK2 RP, 489,076.
PCR assays were carried out in a 25-μl reaction containing the template genomic DNA (250 ng) in 1× polymerase reaction XL buffer II (Perkin–Elmer), 1.1 mM Mg(OAc)2, 200 μM dNTPs, 5 pmol of each primer, and 1 unit of rTth polymerase (Perkin–Elmer). PCR amps were performed in a 2,400 Thermocycler (Perkin–Elmer) with the following temperature profile: an initial denaturation at 94°C for 1 min; 37 cycles (unless otherwise indicated) of denaturation (15 sec at 94°C) and annealing (5 min at 68°C); and final extension at 72°C for 7 min. PCR products were monitored by gel electrophoresis in 1% agarose and by staining with ethidium bromide.
Results and Discussion
Prediction of Rearrangements in the pSym of Rhizobium Sp. NGR234.
Based on the DNA sequence of pNGR234a (9), we localized the major families of direct-repeated sequences (Fig. 1). These families consist of identical elements ranging in size from 2.6 to 4.3 kb. One of these families corresponds to two functional operons (nifHDK1 and nifHDK2) that code for the polypeptides of nitrogenase, the enzyme responsible for the reduction of atmospheric nitrogen. The other four families correspond to insertion sequences. We have used the term amplicon (AMPL) to denote a segment of DNA flanked by two repeated sequences present in direct orientation (4). Recombination between the repeated sequences results in either amp or del of the whole AMPL structure. The DNA families localized in pNGR234a form seven different AMPLs (Fig. 1) ranging in size from 36 to 307 kb.
Figure 1.

Dynamic map of the pSym of Rhizobium sp. NGR234 (pNGR234a). The position and orientation of reiterated DNA sequences are indicated by arrowheads. The origin of replication (oriV) and the first and last bases of the annotation of the sequence as reported previously (9) are indicated. The different AMPLs are represented by arcs outside the plasmid circle. 1, AMPL Rhizobium sp. NGR234 IS5a-b; 2, AMPL Rhizobium sp. NGR234 IS5a-c; 3, AMPL Rhizobium sp. NGR234 IS3b-c; 4, AMPL Rhizobium sp. NGR234 IS5b-c; 5, AMPL Rhizobium sp. NGR234 IS2a-b; 6, AMPL Rhizobium sp. NGR234 IS4b-a; 7, AMPL Rhizobium sp. NGR234 nifHDK1–2.
In a circular replicon, each AMPL might be amplified in two directions, one of them including the origin of replication. However, amp of DNA segments carrying the origin of replication may result in unstable molecules. Actually, after extensive studies in the pSym of Rhizobium etli, we have not detected amps that include the origin of replication (12). Thus, two types of rearrangements are expected from each AMPL: the amp of the region that does not contain the origin of replication, and the del of the same region, giving rise to a shorter replicon containing the origin of replication. This event makes a total of 14 potential rearrangements, mediated by identical direct-reiterated elements in pNGR234a.
Rationale for the Identification of DNA Rearrangements.
To identify the rearrangements predicted from the DNA sequence, we used a PCR-based strategy (Fig. 2). Primers for PCR are synthesized for each of the two repeated sequences that flank an AMPL. The primers must match regions proximal, albeit external, to the homologous repeats. If the FP and the RP of one of the repeats is used for the PCR assay, a product corresponding to the original (wild type) structure of the region will be obtained. DNA rearrangements generate novel recombinant structures that can be revealed by appropriate combinations of primers. The recombinant structure corresponding to a del should be revealed by a PCR product primed by the FP of the initial repeat and the RP of the terminal repeat of the AMPL; that structure corresponding to an amp should be revealed by a PCR product primed by the FP of the terminal repeat and the RP of the initial repeat.
Figure 2.

Identification of genomic rearrangements by PCR. The initial (■) and terminal (□) repeats of an AMPL, and the recombinant structures characteristic of the del (╡) and of the amp (╞) of the AMPL are shown. The position of the PCR primers is shown by arrows. a, FP of the initial repeat; b, RP of the initial repeat; c, FP of the terminal repeat; d, RP of the terminal repeat.
Coincidence Between the Predicted and the Actual Dynamic Map of pNGR234a.
To detect the rearrangements predicted from the DNA sequence of pNGR234a, primers corresponding to regions proximal to each repeated sequence were synthesized. It is important to point out that to ensure the success of the strategy proposed, it is convenient to synthesize and test alternative primers for each position. Our experience indicates that some combinations of primers, although useful in more conventional applications, fail to detect the recombinant structures originated by rearrangements. This may be because of the low concentration of such structures in the wild-type strain (see Unique Rearrangement Profile of Individual Rhizobium Colonies). The exact sequence location of the primers used is indicated in Materials and Methods.
PCR assays were performed with total DNA from the wild-type Rhizobium strain NGR234 as template (Fig. 3, lane A). The PCR products were separated by agarose gel electrophoresis. In all cases, the reactions performed with the FP and RP corresponding to each of the reiterated sequences involved in this study produced large quantities of the expected product (an example is shown in Fig. 3, row 1). Reactions performed with the appropriate combinations of primers to detect the recombinant structures of the 14 predicted rearrangements yielded products of the size expected in all cases (Fig. 3, lane A, rows 2–15). The amount of product obtained varied for the different reactions. DNA from strain ANU265, a derivative of NGR234 cured of the pSym (13), did not yield any of the PCR products (Fig. 3, lane B). To obtain negative controls in the pSym, 10 regions of 2 to 5 kb each were randomly selected and combined in pairs that do not correspond to reiterated sequences; the members of each pair were separated from 30 to 300 kb. For each pair of regions, different combinations of primers that would detect recombinant structures originated by amps or dels were used for PCR. All of the combinations performed failed to detect PCR products derived from the generation of rearrangements (data not shown).
Figure 3.
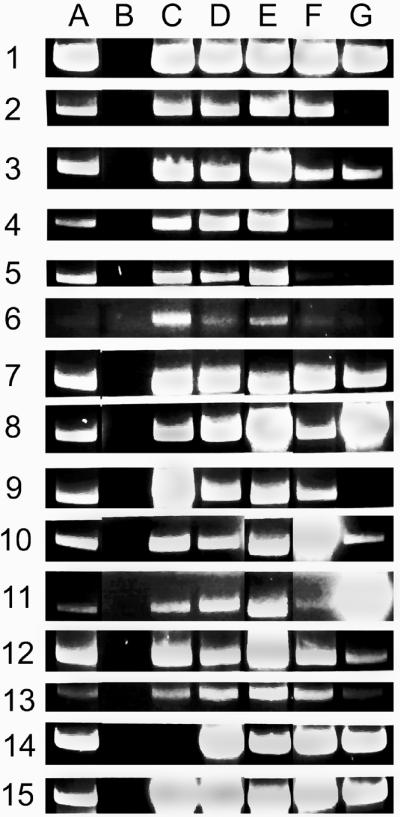
PCR assays in different subpopulations of Rhizobium sp. NGR234. PCR assays were performed by using genomic DNA from different strains as template. The reactions were primed with sets of oligonucleotides that detect different structures. The PCR products were separated by agarose gel electrophoresis and stained with ethidium bromide. The zone of the gel corresponding to each of the expected PCR products is shown. A, wild-type strain; B, ANU265; C, CFNX501; D, CFNX502; E, CFNX503; F, CFNX504; and G, CFNX505. Different pairs of primers were used. For each pair of primers, the size of the PCR product in kb is indicated in the first set of parentheses and the type of structure detected is indicated in the second set of parentheses: I, NGRIS5a FP and RP (6.5) (NGRIS5a); 2, NGRIS5b FP and NGRIS5a RP (6.6) (NGRIS5a-b amp); 3, NGRIS5a FP and NGRIS5b RP (3.1) (NGRIS5a-b del); 4, NGRIS5c FP and NGRIS5a RP (4.1) (NGRIS5a-c amp); 5, NGRIS5a FP and NGRIS5c RP (6.9) (NGRIS5a-c del); 6, NGRIS3c FP and NGRIS3b RP (4.0) (NGRIS3b-c amp); 7, NGRIS3b FP and NGRIS3c RP (4.2) (NGRIS3b-c del); 8, NGRIS5c FP and NGRIS5b RP (3.2) (NGRIS5b-c amp); 9, NGRIS5b FP and NGRIS5c RP (5.8) (NGRIS5b-c del); 10, NGRIS2b FP and NGRIS2a RP (3.7) (NGRIS2a-b amp); 11, NGRIS2a FP and NGRIS2b RP (3.6) (NGRIS2a-b del and NGRIS2b-a amp); 12, NGRIS4a FP and NGRIS4b RP (7.6) (NGRIS4b-a amp); 13, NGRIS4b FP and NGRIS4a RP (7.7) (NGRIS4b-a del); 14, NGRnifHDK2 FP and NGRnifHDK1 RP (4.4) (NGRnifHDK1–2 amp); and 15, NGRnifHDK1 FP and NGRnifHDK2 RP (4.7) (NGRnifHDK1–2 del).
Several of the PCR products that detected the recombinant structures of the rearrangements were analyzed by sequencing about 500 bp of each border and were compared with the DNA sequence reported for pNGR234a. In all cases, the sequence was that expected for the predicted recombinant product (see Characterization of Subpopulations Harboring Specific Rearrangements). These data indicate that the wild-type population contains all of the structures expected from recombination between the members of the different reiterated families, and that the predicted and actual dynamic map of pNGR234a coincide.
Unique Rearrangement Profile of Individual Rhizobium Colonies.
Most probably the proportion of a specific rearrangement in a population is conditioned by the rate of generation of the rearrangement, the reversion rate (in the case of amps), and the relative growth rate of the rearranged cells. In a culture handled for a long time under constant conditions, the rearrangement profile (relative proportion of the different rearrangements) will tend to reach a steady state. If a culture is plated and new cultures are started from single cells, rearrangements will begin to appear, presumably as a stochastic process, as the population grows. By the time when a colony is formed (≈108 cells), the different rearrangements should be present in the population. However, the proportion of each rearrangement will be different, according not only to the rates of generation and reversion of the rearrangements and the relative growth rate of the rearranged cells but, most important, according to the time during which a particular rearrangement first appeared. If a rearrangement appears in the early generations, its concentration in the colony will be larger than if it appears in the late generations. If the rearrangement was already present in a single-plated cell, the colony will be “pure” (see Artificial Selection of Genomic Rearrangements) for such rearrangement.
A culture of the wild-type strain NGR234 was plated, and total DNA from 100 individual colonies was isolated. PCR assays were performed with each DNA using as primers the 14 pairs of oligonucleotides that detect the different rearrangements. Although there was a tendency for some rearrangements to produce higher PCR signals, the overall rearrangement profile was different for each colony (data not shown).
The fact that individual colonies from the same strain present different rearrangement profiles might have biological consequences. Recent data obtained with Escherichia coli populations show a high frequency of chromosomal differences for the repeated fraction of the genome; interestingly, these differences accumulate over time, because almost every individual had a different genetic fingerprint after 10,000 generations (14). Differences in the rearrangement profile could result in a different potentiality for adaptation and evolution of individual cultures derived from the same population.
Artificial Selection of Genomic Rearrangements.
To isolate derivative strains containing specific genomic rearrangements, different genetic procedures have been reported (1, 4). In the present study, we used a strategy that does not involve the introduction of exogenous elements into the genome. It is based on the gradual enrichment of a specific rearrangement in subpopulations of a bacterial strain, without applying any selective pressure. To determine the degree of enrichment of the rearrangement, the recombinant structure characteristic of the rearrangement is monitored by PCR. This enrichment procedure is a case of artificial selection—as opposed to natural selection—because there is not an actual selective pressure to increase the abundance of the corresponding recombinant structures.
To obtain subpopulations of Rhizobium strain NGR234 pure for specific rearrangements, the following protocol produced the best results. The first step consists of plating the wild-type strain to isolate individual colonies. DNA from each colony is purified and used as template for PCR. The colony corresponding to the highest amount of PCR product is selected. In a second step, the selected colony is plated and individual colonies are obtained. Total DNA is isolated from pools of these colonies and is used as template for PCR assays. The pool producing the highest amount of PCR product is selected. Finally, in the third step, DNA from each of the colonies of the pool is used as template for PCR. In a successful experiment, the colony showing the highest amount of PCR product should be pure for the corresponding rearrangement. We are using “purity” as a relative concept, because absolute purity is only reached for certain rearrangements. Actually, a population may be pure in the case of dels, but in the case of amps, 100% purity will never be present because amp is a reversible event. To ascertain the purity of the population, individual colonies are isolated, and the DNA from each colony is used as template for PCR. In the case of dels, all of the DNAs should produce an optimal PCR signal. For amps, the presence of the rearrangement (detected as optimal PCR signals in individual colonies) in the majority of the cells of the population, should be used as criteria of purity. Actually, in the case of amps, a pure subpopulation usually shows optimal PCR signals in >95% of the individual colonies.
The number of colonies and pools that must be screened depends on the relative proportions of cells containing the rearrangement in the wild-type population. In prokaryotes, amps and dels are usually present in frequencies of 10−3 to 10−5 (3, 4). The first step of the protocol described is aimed to increase the proportion of cells containing the rearrangement to about 1 in 103 or higher, which usually is achieved by analyzing 100–300 individual colonies. From this subpopulation, the screening of about 50 pools of 50 colonies each is usually sufficient. If the rearrangement is present at high concentration in the population, the first step may be omitted; in contrast, if the rearrangement is present in very low concentration, the amount of individual colonies in the first step and/or the amount of pools in the second step must be increased.
In this study, we applied the protocol and obtained pure subpopulations corresponding to five different rearrangements: nifHDK1–2 del (strain CFNX501), nifHDK1–2 amp (strain CFNX502), NGRIS5b-c amp (strain CFNX503), NGRIS2a-b amp (strain CFNX504), and an amp that is part of a complex rearrangement described below (strain CFNX505). The rearrangement profile of each strain is shown in Fig. 3. As expected, all of them show an intense PCR signal of the corresponding rearrangement, whereas the rest of the rearrangement profile is unique for each one.
A derivative strain pure for a specific rearrangement represents an appropriate standard to quantify such rearrangement in different populations including the wild type. To this end, we used a procedure based on the amount of PCR product obtained as a function of the number of amp cycles in the reaction. To standardize the quantification procedure, populations of cells that produced positive and negative signals for certain PCR reactions were combined in different proportions; DNA was used as template for PCR, and the amount of product was analyzed at different cycles of the reaction (data not shown). The amount of the PCR product corresponding to specific rearrangements was analyzed at different cycles in both the wild-type and the purified strains (two examples are presented in Fig. 4). From the quantification experiments, we concluded that the wild-type population of NGR234 contains the nifHDK1–2 amp and the nifHDK1–2 del in about 1 in 103 cells, the NGRIS2a-b amp is present in about 1 in 104 cells, and the NGRIS5b-c amp is present in about 1 in 105 cells.
Figure 4.

Quantification of rearrangements in different subpopulations of Rhizobium sp. NGR234. PCR assays were performed for different number of cycles (indicated at the bottom of the lanes). The PCR products were separated by agarose gel electrophoresis and stained with ethidium bromide. DNAs from the wild-type strain and from strains pure for specific rearrangements were used as templates: wild-type strain (A and C); CFNX502 (B); and CFNX503 (D). Different pairs of primers were used. For each pair of primers, the size of the PCR product in kb is indicated in the first set of parentheses and the type of structure detected is indicated in the second set of parentheses: (A and B) NGRnifHDK2 FP and NGRnifHDK1 RP (4.4) (NGRnifHDK1–2 amp); and (C and D) NGRIS5c FP and NGRIS5b RP (3.2) (NGRIS5b-c amp). For quantification, the procedure was standardized as indicated in the text. The relative proportion of cells containing a rearrangement (as indicated in the text) was determined by comparing the number of cycles necessary to synthesize a certain amount of product by using DNA from either the wild type or the subpopulation pure for a specific rearrangement as template.
Characterization of Subpopulations Harboring Specific Rearrangements.
The strains pure for specific rearrangements were further characterized. The DNA sequence of the borders of each PCR product was determined and in all cases shown to be identical to that expected for a recombinant product (data not shown) as compared with the sequence reported for the corresponding regions in pNGR234a (9). Plasmid profiles of the amplified strains (CFNX502, CFNX503, and CFNX504) showed the expected increase in the size of the pSym, whereas the deleted strain (CFNX501) showed a decrease in the size of the plasmid (examples are presented in Fig. 5A).
Figure 5.

Characterization of the pSym of strains CFNX501 and CFNX502. (A) Plasmid profiles stained with ethidium bromide. W and 3, wild-type strain NGR234; 1, CFNX501; 2, CFNX502. The position of the plasmids in the wild-type strain is indicated: a, pNGR234a; b, pNGR234b; c, well entry. (B) HindIII maps. A region of the pSym (from the HindIII site at base 439,970 to the HindIII site at base 501,514) of the wild-type strain NGR234 is shown. The HindIII sites are indicated by arrows and the HindIII fragments are labeled with numbers from 1 to 9. Two small fragments located between fragments labeled 3 and 4 were not taken into account in the analysis. The reiterated sequences nifHDK1 (□) and nifHDK2 (■) are located in HindIII fragments 3 and 7, respectively. The position of the PCR products covering this region is shown below the HindIII map of NGR234; each product is labeled with a number from 81 to 90. The predicted HindIII maps for the corresponding region of strains CFNX501 and CFNX501 also are shown. The new recombinant HindIII fragments produced by the del event in strain CFNX501 (╡) and by the amp event in strain CFNX502 (╞) are labeled X and Y, respectively. (C) Southern blots of total DNA digested with HindIII hybridized against the PCR products covering the region. For each PCR product (indicated at the top), three lines of a Southern blot are shown. The left line corresponds to strain NGR234, the middle line to strain CFNX501, and the right line to strain CFNX502. The position of the corresponding HindIII fragments is indicated.
The complete structure of the pSym of strains CFNX501 and CFNX502 was analyzed. Based on the nucleotide sequence reported (9), the HindIII map of NGR234 and those HindIII patterns corresponding to the predicted structures of the pSym of the rearranged strains were obtained (Fig. 5B). A total of 96 slightly overlapping PCR products of about 6 kb covering the complete sequence of the pSym of strain NGR234 were synthesized (Fig. 5B). Each product was used as probe to hybridize against Southern blots of HindIII-digested DNA of each strain. The hybridization patterns of the rearranged strains were identical to those of NGR234, except for the region involved in the corresponding rearrangement. The rearranged region showed the characteristic features of del or amp: (i) the presence and position of the specific recombinant fragment generated by the rearrangement, and (ii) the absence of hybridization signal of the fragments corresponding to the deleted region (in strain CFNX501) or the increase in intensity of the hybridization signal in the bands corresponding to the fragments included in the amplified region (in strain CFNX502) (Fig. 5C). These experiments confirm the structures predicted for the pSym in these rearranged strains.
Stability of Genomic Rearrangements.
An amplified zone of the genome contains two or more tandem repeats of an AMPL structure. This zone is highly dynamic because of homologous recombination between the tandem repeats. Such events may increase or decrease the level of amp. In the absence of a selective pressure, the amp tends to revert to the wild-type structure.
To test the stability of rearrangements, strains CFNX501, CFNX502, CFNX503, and CFNX504 were grown without selective pressure in both liquid and agar media. Liquid cultures were transferred every day, whereas agar cultures were transferred every second day. At different times, the cultures were plated out and individual colonies were screened for the yield of the corresponding PCR product. After 60 days (≈400 generations) most of the cells (60–80%) of the amplified populations (strains CFNX502, CFNX503, and CFNX504) still contained the rearrangement; as expected, the del present in strain CFNX501 was completely stable (data not shown). Thus, although amplified strains are unstable, they can be handled without selective pressure for relatively long periods. Moreover, by plating the culture and analyzing individual colonies, a new subpopulation, pure for the specific amp, can be obtained.
Multiple Rearrangements and Generation of New AMPLs.
In some cases, a particular rearrangement results in the formation of a new AMPL that was not present in the original genome. A specific case is schematized in Fig. 6. The DNA sequence of pNGR234a shows a region where two overlapping AMPLs are present. One (AMPL A) has NGRIS5b as the initial border and NGRIS5c as the terminal border; the other (AMPL B), which is located inside AMPL A, has NGRIS2a as the initial border and NGRIS2b as the terminal border. From the wild-type NGR234, we obtained strain CFNX503 (see Artificial Selection of Genomic Rearrangements), which is pure for the amp of AMPL A. The structure of the corresponding genomic region of this strain shows that a new AMPL (AMPL C) was formed. It has as initial border an NGRIS2b and as terminal border an NGRIS2a (see Fig. 6). A subpopulation of CFNX503 pure for the amp of AMPL C was searched by the method of artificial selection described above, monitoring with the PCR product obtained by using the FP of NGRIS2a and the RP of NGRIS2b. It should be noted that this pair of primers also could detect a del of AMPL B. One isolate, strain CFNX505, contained both the amp of AMPL A and the amp of AMPL C. Its rearrangement profile is shown in Fig. 3, lane 6. The recombinant structures of the two rearrangements that generated strain CFNX505 were confirmed by sequencing the borders of the corresponding PCR products (data not shown).
Figure 6.

Schematic representation of multiple rearrangements in subpopulations of Rhizobium sp. NGR234. CFNX503 and its derivative CFNX505 are subpopulations pure for specific rearrangements (see text). The position of reiterated sequences (□, NGRIS5b; ■, NGRIS5c; ○, NGRIS2a; and ●, NGRIS2b) of recombinant structures (╡ and ◑) and of PCR primers (1, NGRIS5b FP; 2, NGRIS5b RP; 3, NGRIS5c FP; 4, NGRIS5c RP; a, NGRIS2a FP; b, NGRIS2a RP; c, NGRIS2b FP; and d, NGRIS2b RP) are indicated. A, B, and C correspond to the AMPLs described in the text.
Genomic Design.
The potential for high-frequency rearrangements depends to a great extent on the amount, location, and relative orientation of long-repeated sequences. These elements, coupled to the genomic architecture (number of chromosomes and plasmids, circular and linear replicons) [reviewed by Campbell (15) and Casjens (16)], contribute to shaping the dynamic structure of a genome. Highly dynamic genomes have been suggested for microorganisms that show a distinctively high degree of DNA reiteration such as Rhizobium (17), Streptomyces (18), and Halobacterium (19). Moreover, computer analysis of many sequenced bacterial genomes reveals an unexpected richness in repeated sequences (20).
We have shown in this work that starting with a wild-type population it is possible to isolate a subpopulation pure for a particular rearrangement, and that from such a subpopulation, a new one containing two rearrangements can be obtained. This general strategy can be continued, adding as many new rearrangements as desired.
The analysis of the DNA sequence of a genome offers the possibility of designing pathways of rearrangements leading to different genomic structures. Such structures may, in turn, be obtained by the sequential isolation of subpopulations using the artificial-selection approach proposed. The subpopulations obtained would be natural derivatives of wild-type strains without any introduced exogenous DNA. The strains obtained in this way may be regarded as the product of “natural genetic engineering” processes (21). Such strains may be of value in studies related to genome organization and evolution; some strains could be useful for different applied purposes. In addition, these strategies may provide tools for the analysis of bacteria where conventional genetic analysis is not amenable or convenient, as it happens for many pathogenic bacteria.
Acknowledgments
This paper is dedicated to Jaime Mora. We thank Virginia Quinto, Rosa Ma. Ocampo, and Angeles Moreno for technical assistance. This work was supported in part by grants IN214498 from Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México, and L0013N from Consejo Nacional de Ciencia y Technología, Mexico.
Abbreviations
- pSym
symbiotic plasmid
- AMPL
amplicon
- amp
amplification
- del
deletion
- FP
forward primer
- RP
reverse primer
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Anderson R P, Roth J R. Annu Rev Microbiol. 1977;31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- 2.Petes T D, Hill C W. Annu Rev Genet. 1988;22:147–168. doi: 10.1146/annurev.ge.22.120188.001051. [DOI] [PubMed] [Google Scholar]
- 3.Roth J R, Benson N, Galitski T, Haack K, Lawrence J G, Miesel L. In: Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Neidhardt F C, Curtiss R III, Ingrham J L, Lin E C C, Low K Jr, Magasanik B, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2256–2276. [Google Scholar]
- 4.Romero D, Palacios R. Annu Rev Genet. 1997;31:91–111. doi: 10.1146/annurev.genet.31.1.91. [DOI] [PubMed] [Google Scholar]
- 5.Koonin E V, Mushegian A R. Curr Opin Genet Dev. 1996;6:757–762. doi: 10.1016/s0959-437x(96)80032-3. [DOI] [PubMed] [Google Scholar]
- 6.Lashkari D A, McCusker J H, Davis R W. Proc Natl Acad Sci USA. 1997;94:8945–8947. doi: 10.1073/pnas.94.17.8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pueppke S G, Broughton W J. Mol Plant-Microbe Interact. 1999;12:293–318. doi: 10.1094/MPMI.1999.12.4.293. [DOI] [PubMed] [Google Scholar]
- 8.Flores M, Mavingui P, Girard L, Perret X, Broughton W J, Martínez- Romero E, Dávila G, Palacios R. J Bacteriol. 1998;180:6052–6053. doi: 10.1128/jb.180.22.6052-6053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freiberg C, Fellay R, Bairoch A, Broughton W J, Rosenthal A, Perret X. Nature (London) 1997;387:394–401. doi: 10.1038/387394a0. [DOI] [PubMed] [Google Scholar]
- 10.Hynes M F, McGregor N F. Mol Microbiol. 1990;4:567–574. doi: 10.1111/j.1365-2958.1990.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 11.Flores M, González V, Brom S, Martínez E, Piñero D, Romero D, Dávila G, Palacios R. J Bacteriol. 1987;169:5782–5788. doi: 10.1128/jb.169.12.5782-5788.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romero D, Martínez-Salazar J, Girard L, Brom S, Dávila G, Palacios R, Flores M, Rodríguez C. J Bacteriol. 1995;177:973–980. doi: 10.1128/jb.177.4.973-980.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison N A, Cen Y H, Chen H C, Plazinski J, Ridge R, Rolfe B G. J Bacteriol. 1984;160:483–487. doi: 10.1128/jb.160.1.483-487.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papadopoulos D, Schneider D, Meier-Eiss J, Arber W, Lenski R E, Blot M. Proc Natl Acad Sci USA. 1999;96:3807–3812. doi: 10.1073/pnas.96.7.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell A M. Curr Opin Genet Dev. 1993;3:837–844. doi: 10.1016/0959-437x(93)90002-7. [DOI] [PubMed] [Google Scholar]
- 16.Casjens S. Annu Rev Genet. 1998;32:339–377. doi: 10.1146/annurev.genet.32.1.339. [DOI] [PubMed] [Google Scholar]
- 17.Flores M, González V, Pardo M A, Leija A, Martínez E, Romero D, Piñero D, Dávila G, Palacios R. J Bacteriol. 1988;170:1191–1196. doi: 10.1128/jb.170.3.1191-1196.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopwood D A, Kieser T. In: The Bacterial Chromosome. Drlica K, Riley M, editors. Washington, DC: Am. Soc. Microbiol.; 1990. pp. 147–161. [Google Scholar]
- 19.Sapienza C, Rose M R, Doolittle W F. Nature (London) 1982;299:182–185. doi: 10.1038/299182a0. [DOI] [PubMed] [Google Scholar]
- 20.Romero D, Martínez-Salazar J, Ortiz E, Rodríguez C, Valencia Morales E. Res Microbiol. 1999;150:735–743. doi: 10.1016/s0923-2508(99)00119-9. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro J A. Ann NY Acad Sci. 1999;870:23–35. doi: 10.1111/j.1749-6632.1999.tb08862.x. [DOI] [PubMed] [Google Scholar]