

Abstract
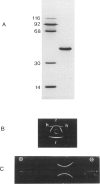
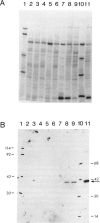
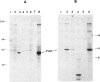
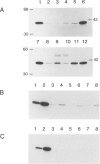
Hereditary tyrosinemia is characterized by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH; E.C.3.7.1.2), the last enzyme in the catabolic pathway of tyrosine. FAH was purified from rat and human liver and was used to immunize rabbits. Specific antibodies were used to probe protein extracts of livers and other tissues of normal and tyrosinemic patients. No immunoreactive FAH band was observed on immunoblots of liver, kidneys, and lymphocytes from patients presenting with the acute form of hereditary tyrosinemia. Patients with the chronic form had immunoreactive FAH at a level approximately 20% of normal liver values, which was correlated with the measured enzymatic activity. Immunoblot analysis of aborted fetal tissues revealed normal FAH immunoreactivity in normal liver and kidneys. No FAH immunoreactivity was found in liver and kidneys of tyrosinemic fetuses. The presence of FAH immunoreactivity in normal fetal tissues suggests that deficient FAH activity in tyrosinemia is not simply related to a developmentally regulated expression of the enzyme. By this immunoblot assay, FAH was detected in most human tissues, with maximal immunoreactivity in liver and kidneys and with only trace amounts in chorionic villi and cultured amniocytes. These data confirm that the primary defect in the acute form of hereditary tyrosinemia is an absence of FAH. Moreover, these data suggest that both clinical forms of the disease have a different molecular basis.
Full text
PDF

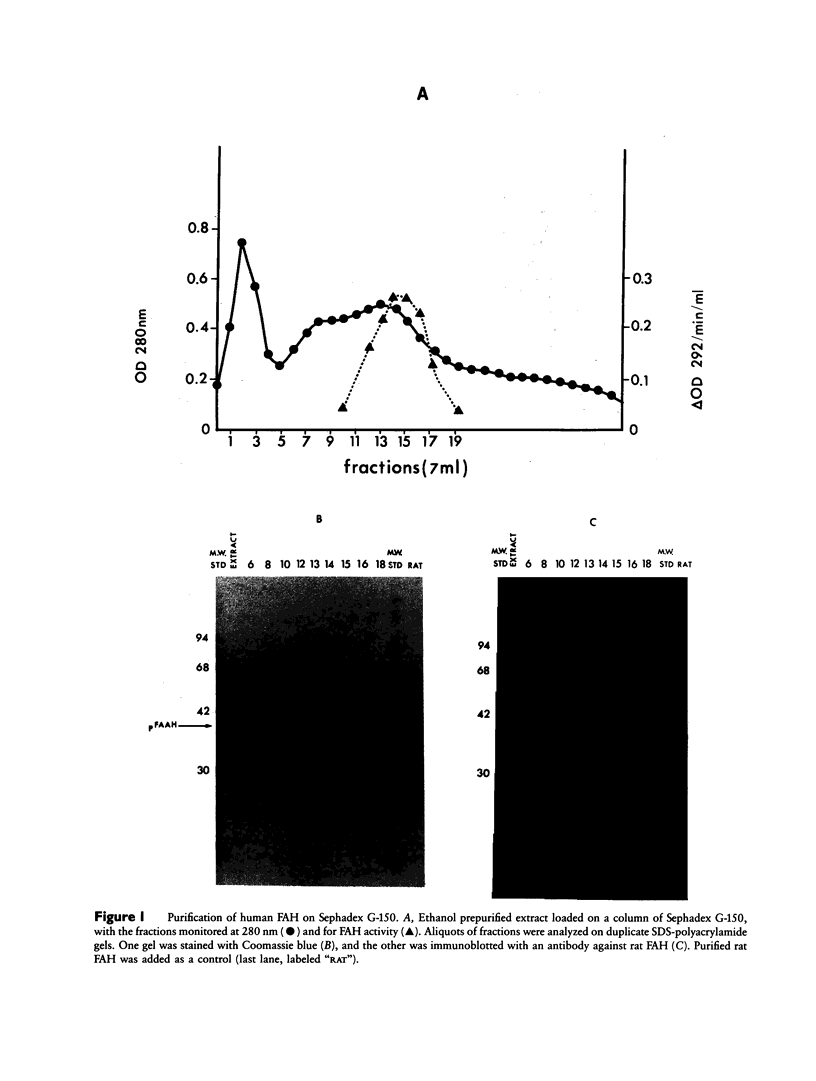
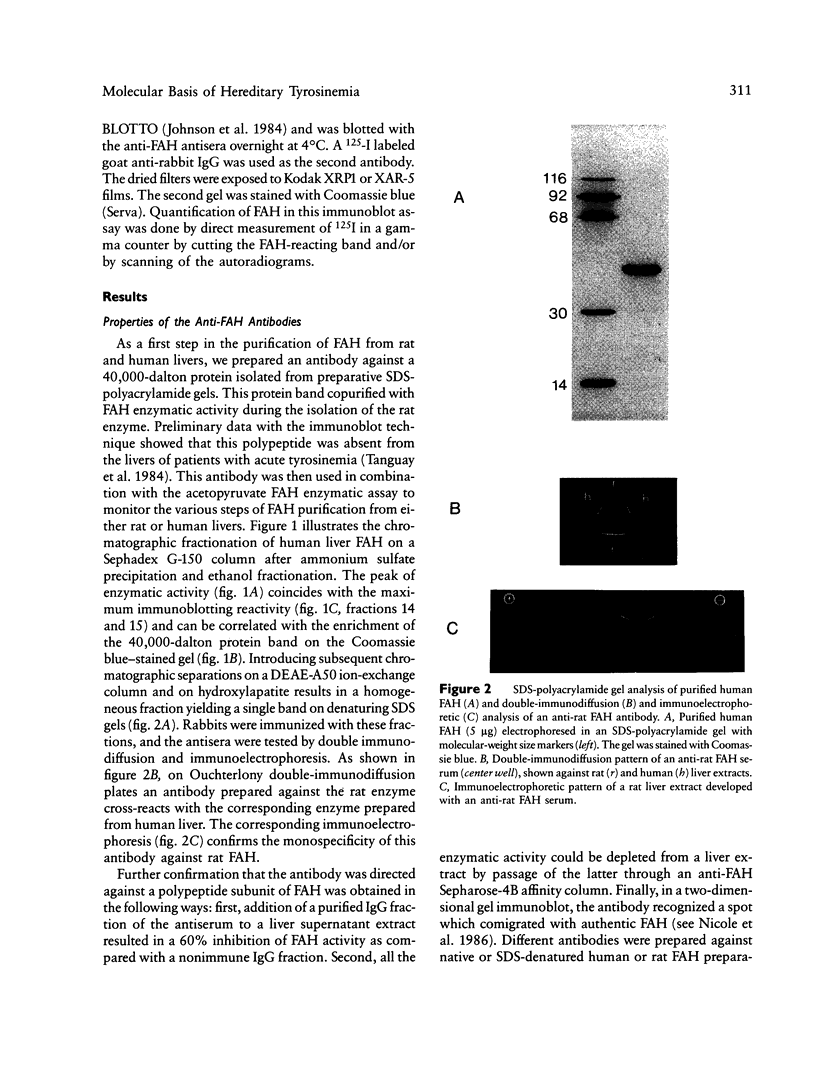

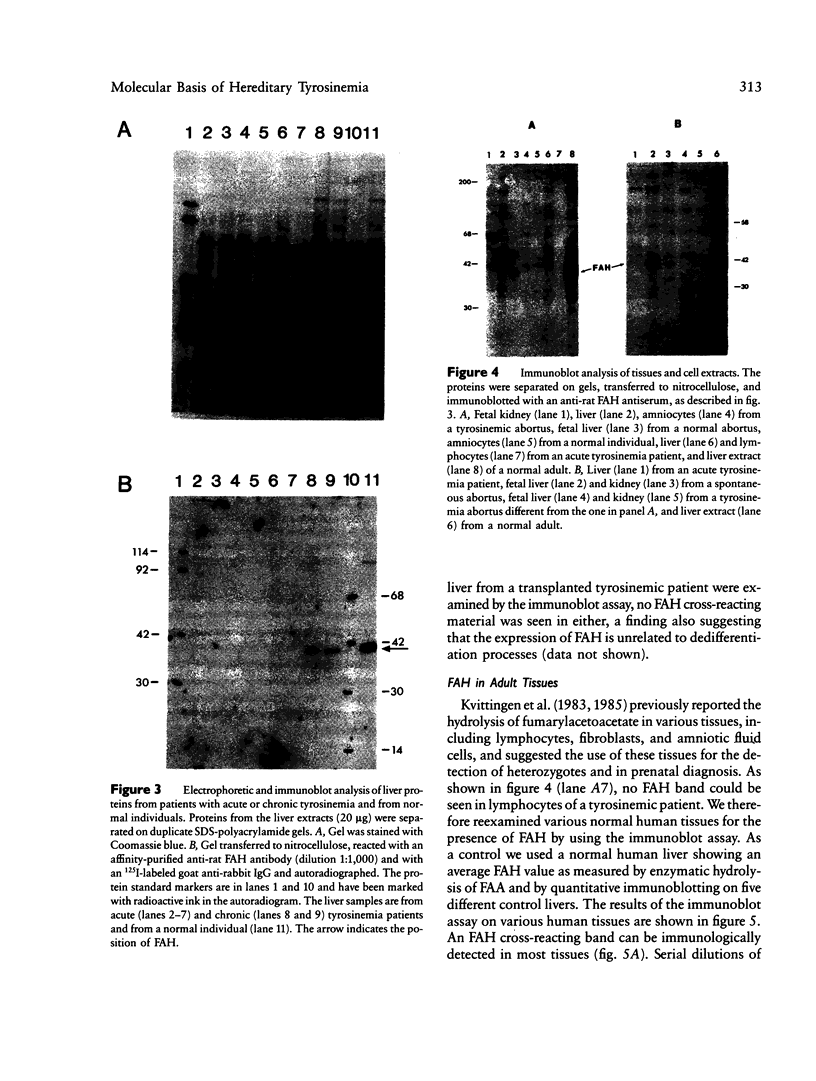



Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Berger R., Smit G. P., Stoker-de Vries S. A., Duran M., Ketting D., Wadman S. K. Deficiency of fumarylacetoacetase in a patient with hereditary tyrosinemia. Clin Chim Acta. 1981 Jul 18;114(1):37–44. doi: 10.1016/0009-8981(81)90225-4. [DOI] [PubMed] [Google Scholar]
- Berger R., Van Faassen H., Taanman J. W., De Vries H., Agsteribbe E. Type I tyrosinemia: lack of immunologically detectable fumarylacetoacetase enzyme protein in tissues and cell extracts. Pediatr Res. 1987 Oct;22(4):394–398. doi: 10.1203/00006450-198710000-00005. [DOI] [PubMed] [Google Scholar]
- Bergeron P., Laberge C., Grenier A. Hereditary tyrosinemia in the province of Quebec: prevalence at birth and geographic distribution. Clin Genet. 1974;5(2):157–162. doi: 10.1111/j.1399-0004.1974.tb01677.x. [DOI] [PubMed] [Google Scholar]
- Furukawa N., Kinugasa A., Seo T., Ishii T., Ota T., Machida Y., Inoue F., Imashuku S., Kusunoki T., Takamatsu T. Enzyme defect in a case of tyrosinemia type I, acute form. Pediatr Res. 1984 May;18(5):463–466. doi: 10.1203/00006450-198405000-00014. [DOI] [PubMed] [Google Scholar]
- Gagné R., Lescault A., Grenier A., Laberge C., Mélançon S. B., Dallaire L. Prenatal diagnosis of hereditary tyrosinaemia: measurement of succinylacetone in amniotic fluid. Prenat Diagn. 1982 Jul;2(3):185–188. doi: 10.1002/pd.1970020307. [DOI] [PubMed] [Google Scholar]
- Gray R. G., Patrick A. D., Preston F. E., Whitfield M. F. Acute hereditary tyrosinaemia type I: clinical, biochemical and haematological studies in twins. J Inherit Metab Dis. 1981;4(1):37–40. doi: 10.1007/BF02263580. [DOI] [PubMed] [Google Scholar]
- Holme E., Lindblad B., Lindstedt S. Possibilities for treatment and for early prenatal diagnosis of hereditary tyrosinaemia. Lancet. 1985 Mar 2;1(8427):527–527. doi: 10.1016/s0140-6736(85)92132-4. [DOI] [PubMed] [Google Scholar]
- Hsiang H. H., Sim S. S., Mahuran D. J., Schmidt D. E., Jr Purification and properties of a diketo acid hydrolase from beef liver. Biochemistry. 1972 May 23;11(11):2098–2102. doi: 10.1021/bi00761a016. [DOI] [PubMed] [Google Scholar]
- Kvittingen E. A., Halvorsen S., Jellum E. Deficient fumarylacetoacetate fumarylhydrolase activity in lymphocytes and fibroblasts from patients with hereditary tyrosinemia. Pediatr Res. 1983 Jul;17(7):541–544. doi: 10.1203/00006450-198307000-00005. [DOI] [PubMed] [Google Scholar]
- Kvittingen E. A. Hereditary tyrosinemia type I--an overview. Scand J Clin Lab Invest Suppl. 1986;184:27–34. [PubMed] [Google Scholar]
- Kvittingen E. A., Jellum E., Stokke O. Assay of fumarylacetoacetate fumarylhydrolase in human liver-deficient activity in a case of hereditary tyrosinemia. Clin Chim Acta. 1981 Sep;115(3):311–319. doi: 10.1016/0009-8981(81)90244-8. [DOI] [PubMed] [Google Scholar]
- Kvittingen E. A., Steinmann B., Gitzelmann R., Leonard J. V., Andria G., Børresen A. L., Mossman J., Micara G., Lindblad B. Prenatal diagnosis of hereditary tyrosinemia by determination of fumarylacetoacetase in cultured amniotic fluid cells. Pediatr Res. 1985 Apr;19(4):334–337. doi: 10.1203/00006450-198519040-00002. [DOI] [PubMed] [Google Scholar]
- Laberge C. Hereditary tyrosinemia in a French Canadian isolate. Am J Hum Genet. 1969 Jan;21(1):36–45. [PMC free article] [PubMed] [Google Scholar]
- Larochelle J., Mortezai A., Belanger M., Tremblay M., Claveau J. C., Aubin G. Experience with 37 infants with tyrosinemia. Can Med Assoc J. 1967 Oct 28;97(18):1051–1054. [PMC free article] [PubMed] [Google Scholar]
- Lindblad B., Lindstedt S., Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci U S A. 1977 Oct;74(10):4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicole L. M., Valet J. P., Laberge C., Tanguay R. M. Purification of mRNA coding for the enzyme deficient in hereditary tyrosinemia, fumarylacetoacetate hydrolase. Biochem Cell Biol. 1986 May;64(5):489–493. doi: 10.1139/o86-068. [DOI] [PubMed] [Google Scholar]
- O'Farrell P. H. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975 May 25;250(10):4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Sarkar G., Sommer S. S. Access to a messenger RNA sequence or its protein product is not limited by tissue or species specificity. Science. 1989 Apr 21;244(4902):331–334. doi: 10.1126/science.2565599. [DOI] [PubMed] [Google Scholar]
- Thomas J. O., Kornberg R. D. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci U S A. 1975 Jul;72(7):2626–2630. doi: 10.1073/pnas.72.7.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979 Sep;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg A. G., Mize C. E., Worthen H. G. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr. 1976 Mar;88(3):434–438. doi: 10.1016/s0022-3476(76)80259-4. [DOI] [PubMed] [Google Scholar]