

Abstract
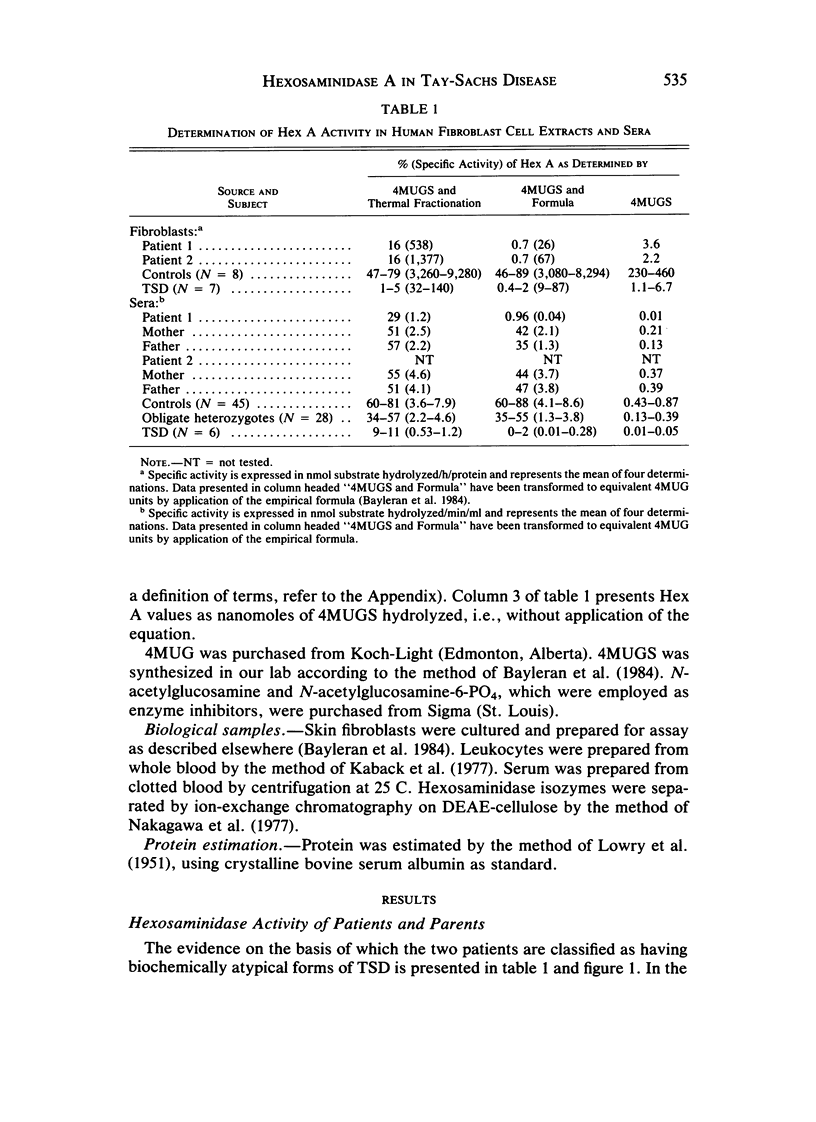
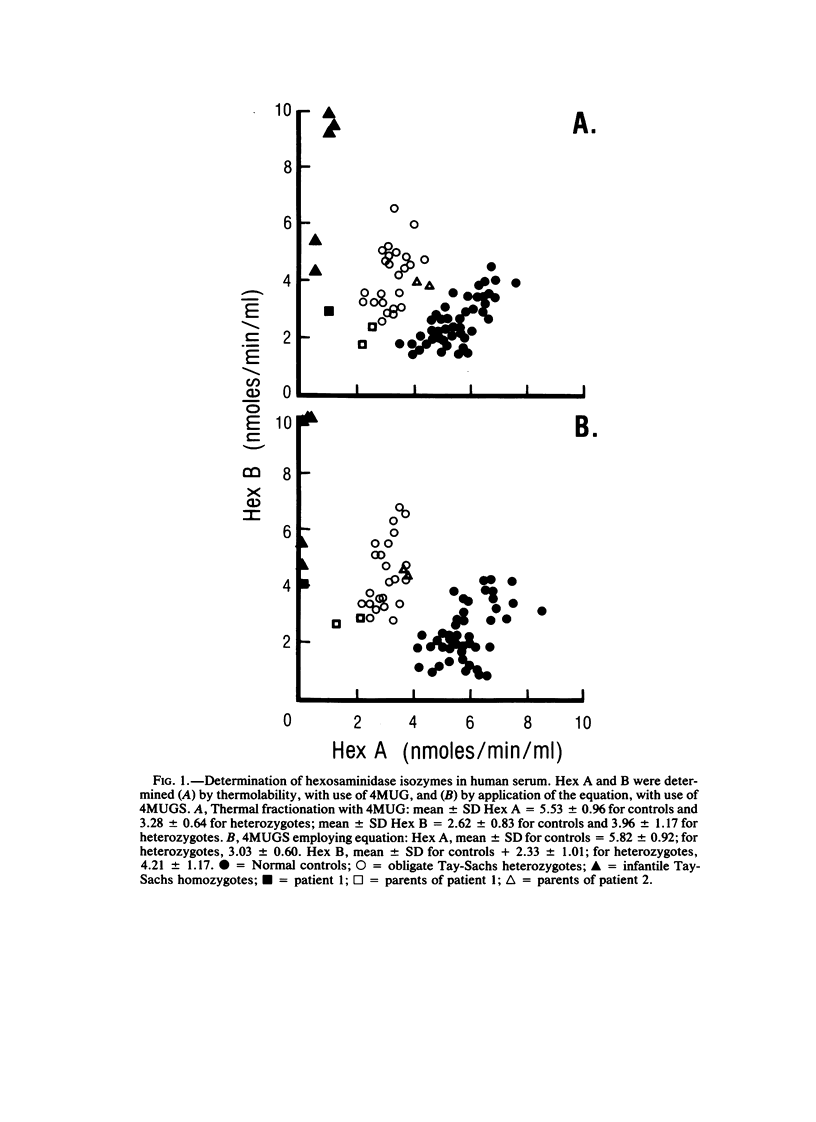
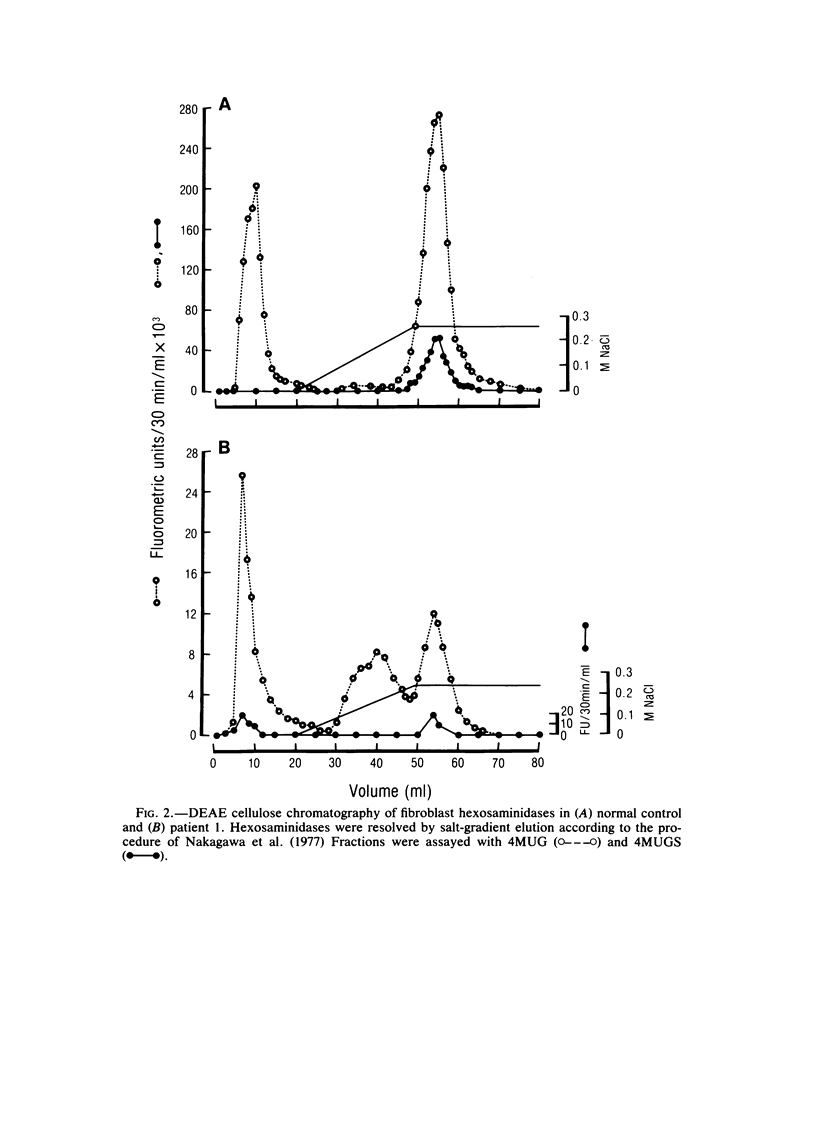
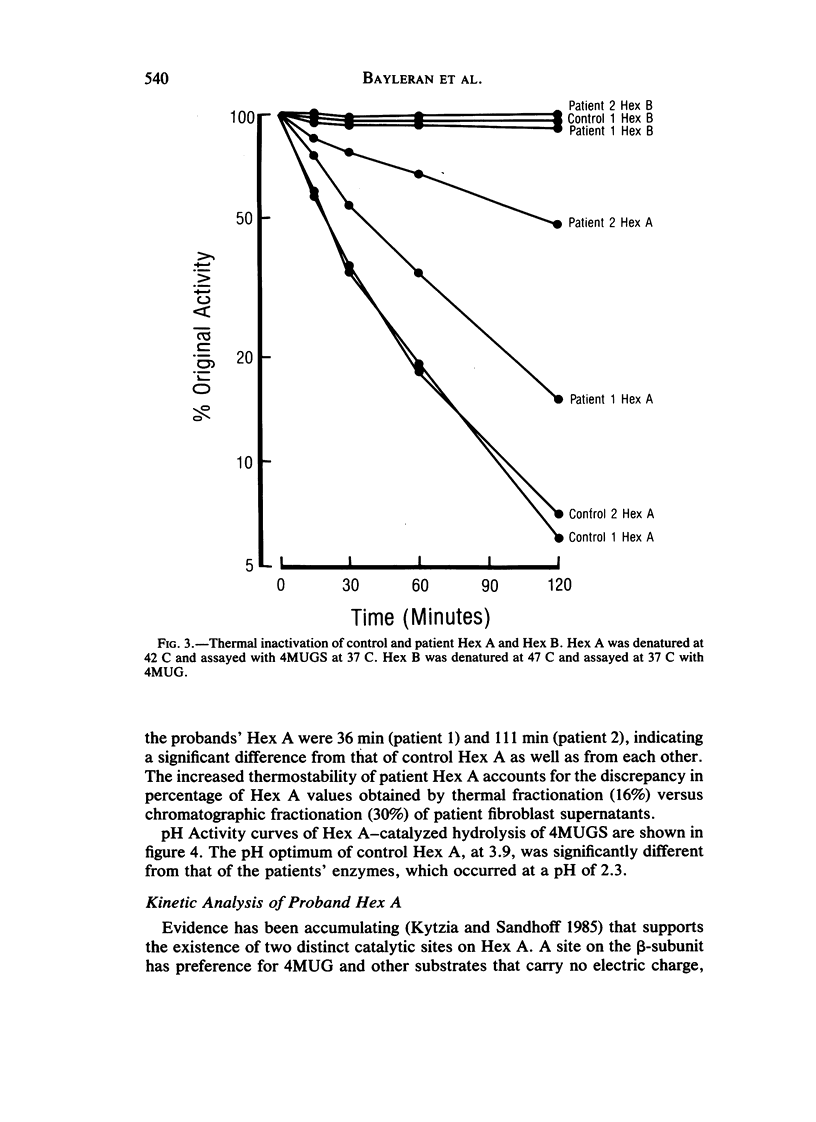
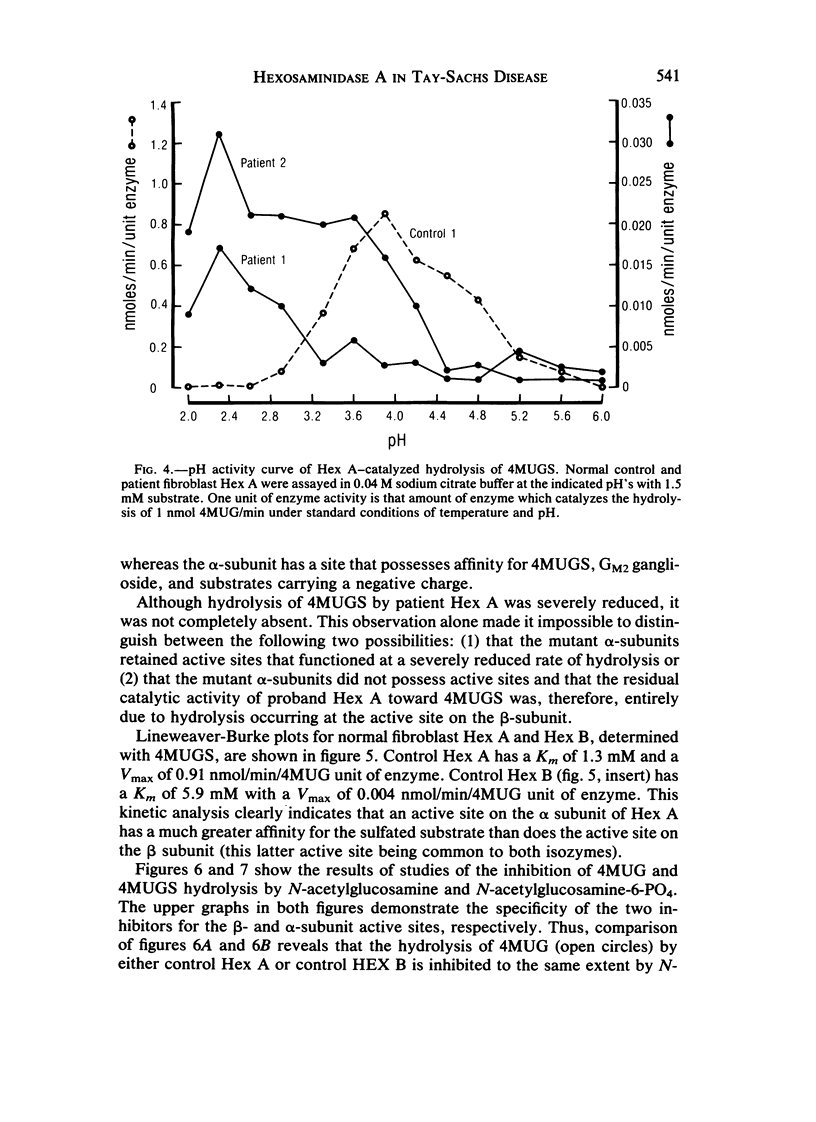
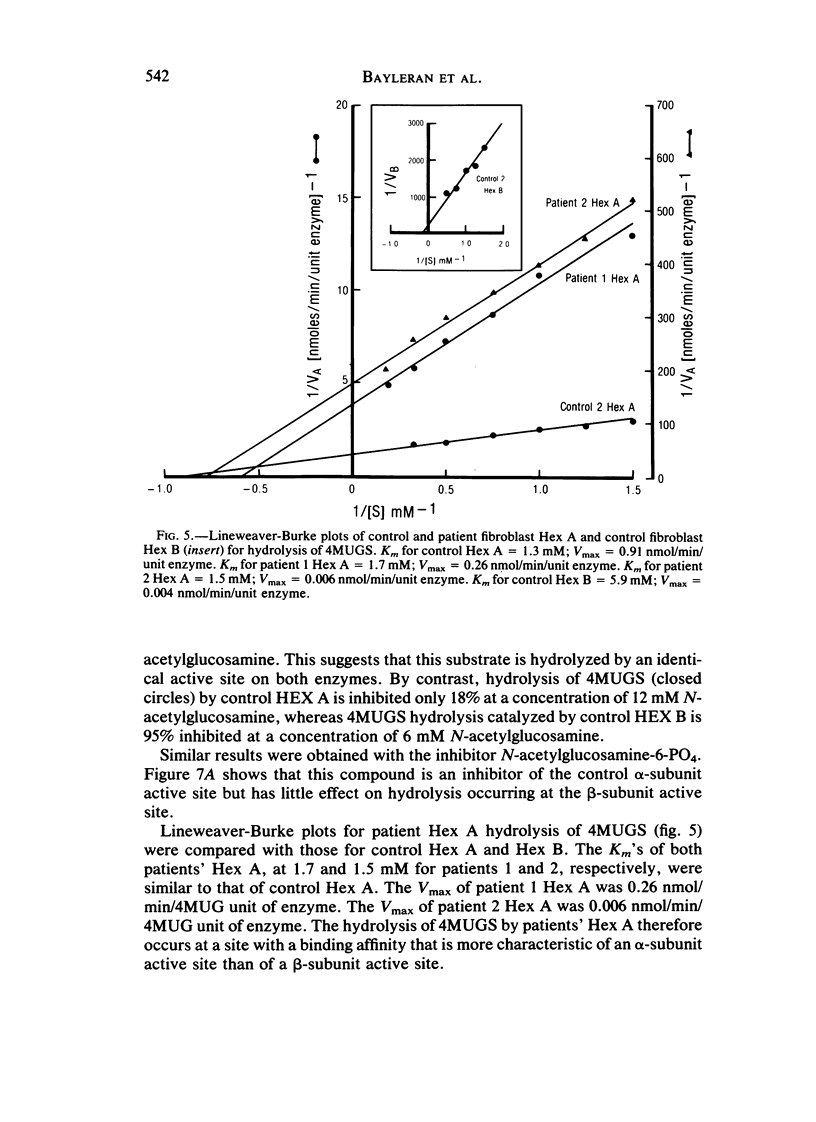
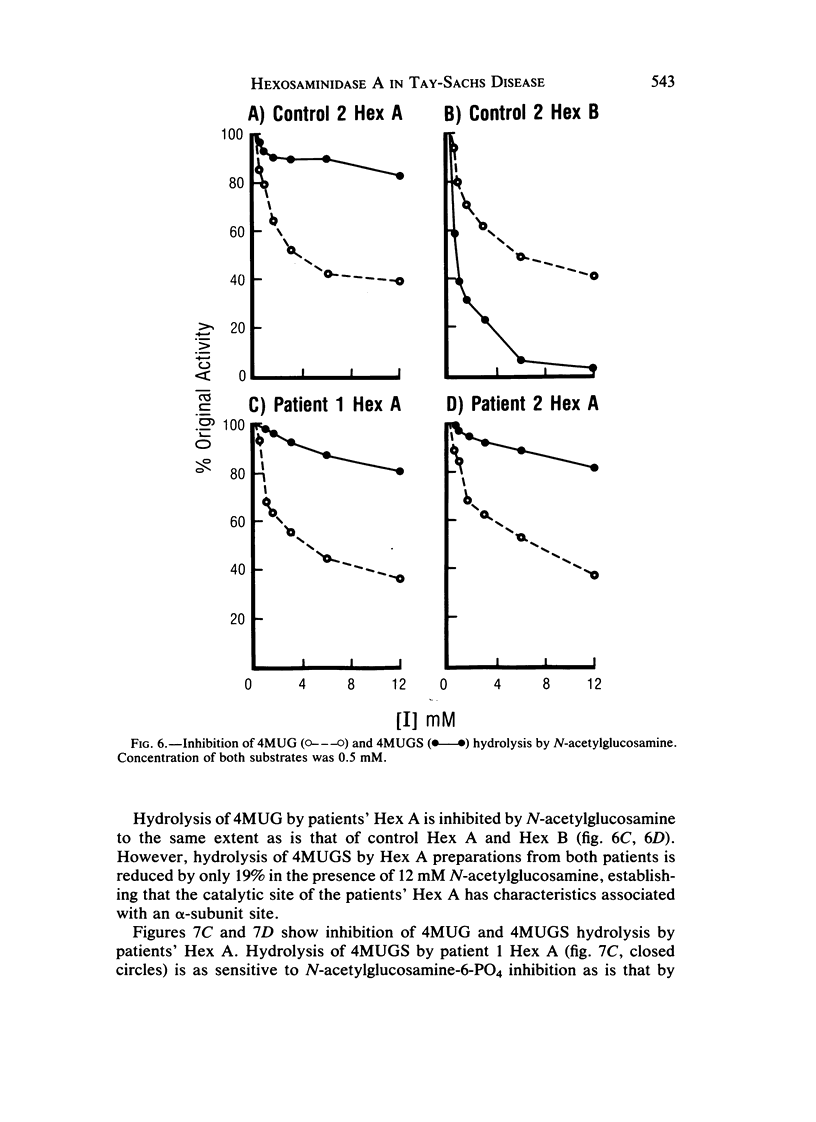
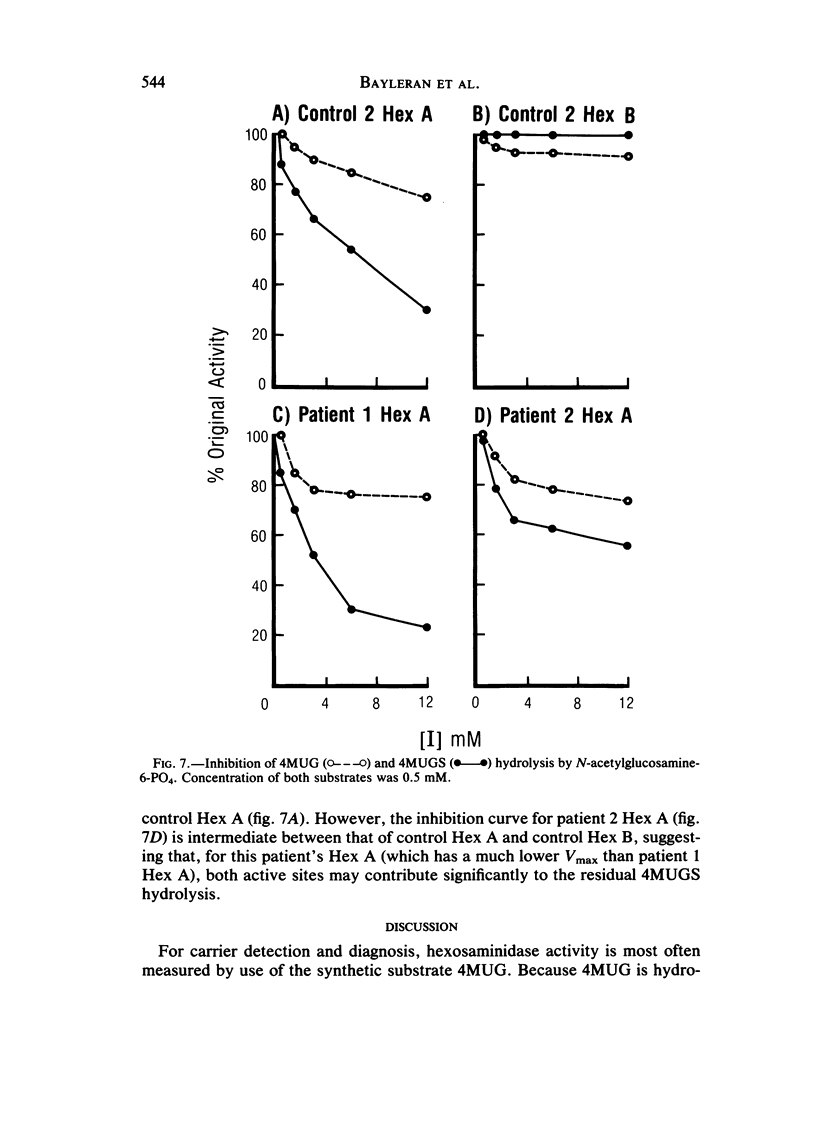
Cases of infantile Tay-Sachs disease (TSD) with high residual hexosaminidase A (Hex A) activity have recently been described. The clinical presentation of the disease in these patients is identical to that found among Ashkenazi-Jewish patients. Fibroblasts from two such TSD patients had Hex A activity comprising 16% of total Hex when measured by thermal fractionation and quantitation with 4-methylumbelliferyl-beta-D-N-acetylglucosamine (4MUG). Hydrolysis of 4-methylumbelliferyl-beta-D-N-acetylglucosamine-6-SO4 (4MUGS) by patient fibroblast extracts is catalyzed by an enzyme activity that comprises less than 1% of total Hex. Kinetic analysis of patient Hex A by using 4MUGS revealed Km's similar to that of control Hex A but Vmax's significantly different from that of the control enzyme. The inhibitors N-acetylglucosamine and N-acetylglucosamine-6-PO4 were used to distinguish between active sites associated with the two different subunits of Hex A. A beta-subunit site with little activity toward 4MUGS is sensitive to N-acetylglucosamine but resistant to N-acetylglucosamine-6-PO4. This site accounts for most of the hydrolysis of 4MUG. By contrast, an alpha-subunit site that is sensitive to N-acetylglucosamine-6-PO4 but resistant to N-acetylglucosamine accounts for almost all of the hydrolysis of 4MUGS. In mutant cells, this site retains the ability to bind substrate but is deficient in catalytic activity toward 4MUGS. The pH optima of patients' Hex A is shifted to a more acidic range, and the enzymes are significantly more thermostable than control Hex A. By using the thermal fractionation procedure for serum isozyme discrimination, one parent of each patient is unambiguously classified as heterozygous for the TSD gene whereas the other parent has test values in the grey zone. When parents are tested by use of 4MUGS, however, all four parents are classified as heterozygotes. Comparison of the results of both assay procedures allows the carrier of the atypical TSD allele to be recognized and identifies the probands as compound heterozygotes.
Full text
PDF








Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Andermann E., Scriver C. R., Wolfe L. S., Dansky L., Andermann F. Genetic variants of Tay-Sachs disease: Tay-Sachs disease and Sandhoff's disease in French Canadians, juvenile Tay-Sachs disease in Lebanese Canadians, and a Tay-Sachs screening program in the French-Canadian population. Prog Clin Biol Res. 1977;18:161–188. [PubMed] [Google Scholar]
- Bayleran J., Hechtman P., Saray W. Synthesis of 4-methylumbelliferyl-beta-D-N-acetylglucosamine-6-sulfate and its use in classification of GM2 gangliosidosis genotypes. Clin Chim Acta. 1984 Nov 15;143(2):73–89. doi: 10.1016/0009-8981(84)90215-8. [DOI] [PubMed] [Google Scholar]
- Ben-Yoseph Y., Reid J. E., Shapiro B., Nadler H. L. Diagnosis and carrier detection of Tay-Sachs disease: direct determination of hexosaminidase A using 4-methylumbelliferyl derivatives of beta-N-acetylglucosamine-6-sulfate and beta-N-acetylgalactosamine-6-sulfate. Am J Hum Genet. 1985 Jul;37(4):733–740. [PMC free article] [PubMed] [Google Scholar]
- Burg J., Conzelmann E., Sandhoff K., Solomon E., Swallow D. M. Mapping of the gene coding for the human GM2 activator protein to chromosome 5. Ann Hum Genet. 1985 Jan;49(Pt 1):41–45. doi: 10.1111/j.1469-1809.1985.tb01674.x. [DOI] [PubMed] [Google Scholar]
- Charrow J., Inui K., Wenger D. A. Late onset GM2 gangliosidosis: an alpha-locus genetic compound with near normal hexosaminidase activity. Clin Genet. 1985 Jan;27(1):78–84. doi: 10.1111/j.1399-0004.1985.tb00188.x. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Kytzia H. J., Navon R., Sandhoff K. Ganglioside GM2 N-acetyl-beta-D-galactosaminidase activity in cultured fibroblasts of late-infantile and adult GM2 gangliosidosis patients and of healthy probands with low hexosaminidase level. Am J Hum Genet. 1983 Sep;35(5):900–913. [PMC free article] [PubMed] [Google Scholar]
- Conzelmann E., Nehrkorn H., Kytzia H. J., Sandhoff K., Macek M., Lehovský M., Elleder M., Jirásek A., Kobilková J. Prenatal diagnosis of GM2 gangliosidosis with high residual hexosaminidase A activity (variant B1; pseudo AB variant). Pediatr Res. 1985 Nov;19(11):1220–1224. doi: 10.1203/00006450-198511000-00022. [DOI] [PubMed] [Google Scholar]
- Fuchs W., Navon R., Kaback M. M., Kresse H. Tay-Sachs disease: one-step assay of beta-N-acetylhexosaminidase in serum with a sulphated chromogenic substrate. Clin Chim Acta. 1983 Oct 14;133(3):253–261. doi: 10.1016/0009-8981(83)90269-3. [DOI] [PubMed] [Google Scholar]
- Geiger B., Arnon R. Chemical characterization and subunit structure of human N-acetylhexosaminidases A and B. Biochemistry. 1976 Aug 10;15(16):3484–3493. doi: 10.1021/bi00661a014. [DOI] [PubMed] [Google Scholar]
- Goldman J. E., Yamanaka T., Rapin I., Adachi M., Suzuki K., Suzuki K. The AB-variant of GM2-gangliosidosis. Clinical, biochemical, and pathological studies of two patients. Acta Neuropathol. 1980;52(3):189–202. doi: 10.1007/BF00705807. [DOI] [PubMed] [Google Scholar]
- Hechtman P., Kachra Z. Interaction of activating protein and surfactants with human liver hexosaminidase A and GM2 ganglioside. Biochem J. 1980 Mar 1;185(3):583–591. doi: 10.1042/bj1850583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hechtman P., Khoo K., Isaacs C. A new form of residual hexosaminidase activity in infantile Tay Sachs disease fibroblasts. Clin Genet. 1983 Sep;24(3):206–215. doi: 10.1111/j.1399-0004.1983.tb02241.x. [DOI] [PubMed] [Google Scholar]
- Inui K., Grebner E. E., Jackson L. G., Wenger D. A. Juvenile GM2 gangliosidosis (AMB variant): inability to activate hexosaminidase A by activator protein. Am J Hum Genet. 1983 Jul;35(4):551–564. [PMC free article] [PubMed] [Google Scholar]
- Kytzia H. J., Hinrichs U., Maire I., Suzuki K., Sandhoff K. Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity. EMBO J. 1983;2(7):1201–1205. doi: 10.1002/j.1460-2075.1983.tb01567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kytzia H. J., Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985 Jun 25;260(12):7568–7572. [PubMed] [Google Scholar]
- LEABACK D. H., WALKER P. G. Studies on glucosaminidase. 4. The fluorimetric assay of N-acetyl-beta-glucosaminidase. Biochem J. 1961 Jan;78:151–156. doi: 10.1042/bj0780151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Li S. C., Hirabayashi Y., Li Y. T. A new variant of type-AB GM2-gangliosidosis. Biochem Biophys Res Commun. 1981 Jul 30;101(2):479–485. doi: 10.1016/0006-291x(81)91285-7. [DOI] [PubMed] [Google Scholar]
- Myerowitz R., Hogikyan N. D. Different mutations in Ashkenazi Jewish and non-Jewish French Canadians with Tay-Sachs disease. Science. 1986 Jun 27;232(4758):1646–1648. doi: 10.1126/science.3754980. [DOI] [PubMed] [Google Scholar]
- Nakagawa S., Kumin S., Nitowsky H. M. Human hexosaminidase isozymes: chromatographic separation as an aid to heterozygote identification. Clin Chim Acta. 1977 Mar 1;75(2):181–191. doi: 10.1016/0009-8981(77)90189-9. [DOI] [PubMed] [Google Scholar]
- Proia R. L., Neufeld E. F. Synthesis of beta-hexosaminidase in cell-free translation and in intact fibroblasts: an insoluble precursor alpha chain in a rare form of Tay-Sachs disease. Proc Natl Acad Sci U S A. 1982 Oct;79(20):6360–6364. doi: 10.1073/pnas.79.20.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderfeld S., Brendler S., Sandhoff K., Galjaard H., Hoogeveen A. T. Genetic complementation in somatic cell hybrids of four variants of infantile GM2 gangliosidosis. Hum Genet. 1985;71(3):196–200. doi: 10.1007/BF00284572. [DOI] [PubMed] [Google Scholar]