

Abstract
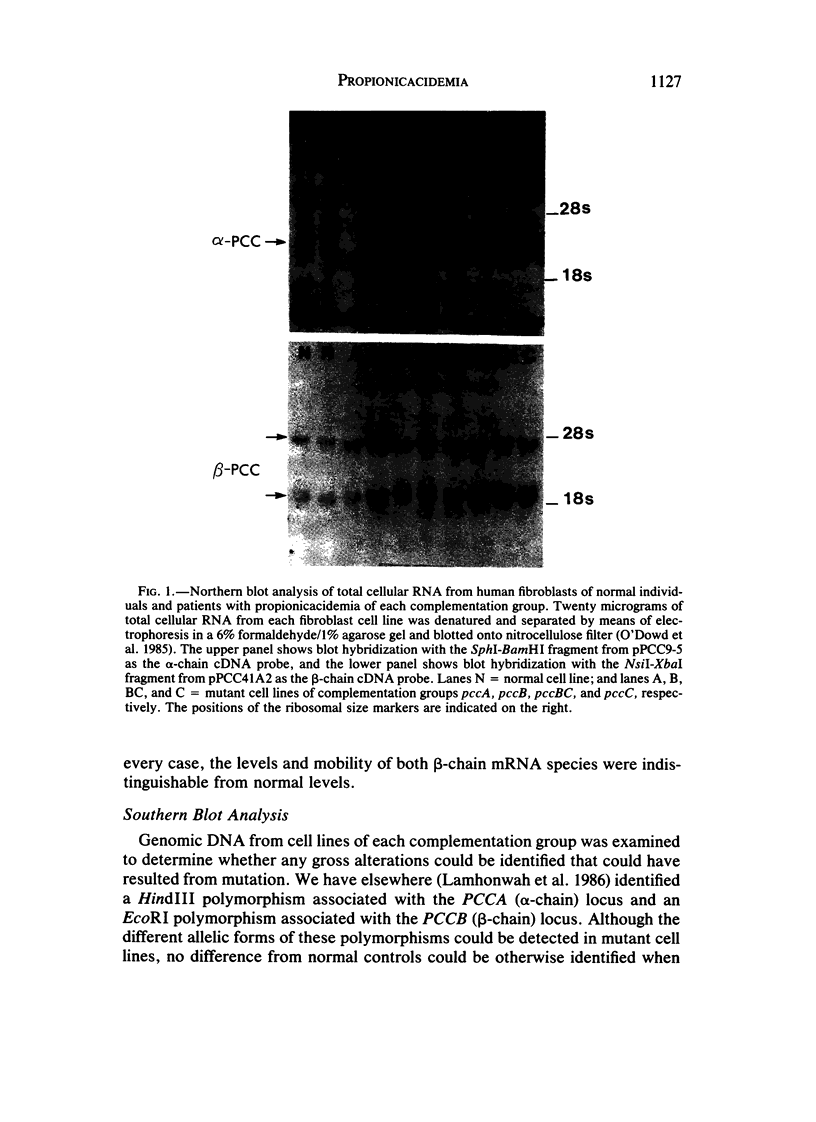
Propionicacidemia is an autosomal recessive metabolic disease resulting from a deficiency of propionyl-CoA carboxylase (PCC) activity. The enzyme has the structure alpha 4 beta 4, with the alpha chain containing a covalently bound biotin prosthetic group. Patients have been placed into two major complementation groups, pccA and pccBC, that may correspond to the genes encoding the alpha and beta chains of PCC. The pccBC group is further divided into two subgroups, pccB and pccC, apparently owing to intragenic complementation. We previously reported combined alpha- and beta-chain deficiency in pccA mutants and absence of beta chain in pccC and pccBC mutants after isotope-tracer labeling and immunoprecipitation of cultured-fibroblast extracts. Using cDNA clones coding for the alpha and beta chains as probes, we found absence of alpha mRNA in four of six pccA strains and presence of beta mRNA in all pccA mutants studied. We also found presence of both alpha and beta mRNAs in three pccBC, two pccB, and three pccC mutants. From these data, we confirm the gene assignments of the complementation groups (PCCA gene = pccA complementation group; PCCB gene = pccBC and subgroups) and support the view that pccA patients synthesize a normal beta chain that is rapidly degraded in the absence of complexing with alpha chains.
Full text
PDF






Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Ando T., Rasmussen K., Nyhan W. L., Donnell G. N., Barnes N. D. Propionic acidemia in patients with ketotic hyperglycinemia. J Pediatr. 1971 May;78(5):827–832. doi: 10.1016/s0022-3476(71)80354-2. [DOI] [PubMed] [Google Scholar]
- Brandt I. K., Hsia Y. E., Clement D. H., Provence S. A. Propionicacidemia (ketotic hyperglycinemia): dietary treatment resulting in normal growth and development. Pediatrics. 1974 Mar;53(3):391–395. [PubMed] [Google Scholar]
- DULBECCO R., VOGT M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J Exp Med. 1954 Feb;99(2):167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Mahuran D., Kronis A. Purification of human liver propionyl-CoA carboxylase by carbon tetrachloride extraction and monomeric avidin affinity chromatography. Arch Biochem Biophys. 1980 May;201(2):669–673. doi: 10.1016/0003-9861(80)90557-3. [DOI] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Scully K. J., Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977 Jul;29(4):378–388. [PMC free article] [PubMed] [Google Scholar]
- Hsia Y. E., Scully K. J., Rosenberg L. E. Defective propionate carboxylation in ketotic hyperglycinaemia. Lancet. 1969 Apr 12;1(7598):757–758. doi: 10.1016/s0140-6736(69)91757-7. [DOI] [PubMed] [Google Scholar]
- Lam Hon Wah A. M., Lam K. F., Tsui F., Robinson B., Saunders M. E., Gravel R. A. Assignment of the alpha and beta chains of human propionyl-CoA carboxylase to genetic complementation groups. Am J Hum Genet. 1983 Sep;35(5):889–899. [PMC free article] [PubMed] [Google Scholar]
- Lamhonwah A. M., Barankiewicz T. J., Willard H. F., Mahuran D. J., Quan F., Gravel R. A. Isolation of cDNA clones coding for the alpha and beta chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc Natl Acad Sci U S A. 1986 Jul;83(13):4864–4868. doi: 10.1073/pnas.83.13.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dowd B. F., Quan F., Willard H. F., Lamhonwah A. M., Korneluk R. G., Lowden J. A., Gravel R. A., Mahuran D. J. Isolation of cDNA clones coding for the beta subunit of human beta-hexosaminidase. Proc Natl Acad Sci U S A. 1985 Feb;82(4):1184–1188. doi: 10.1073/pnas.82.4.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M., Sweetman L., Robinson B., Roth K., Cohn R., Gravel R. A. Biotin-response organicaciduria. Multiple carboxylase defects and complementation studies with propionicacidemia in cultured fibroblasts. J Clin Invest. 1979 Dec;64(6):1695–1702. doi: 10.1172/JCI109632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975 Nov 5;98(3):503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- Willard H. F., Smith K. D., Sutherland J. Isolation and characterization of a major tandem repeat family from the human X chromosome. Nucleic Acids Res. 1983 Apr 11;11(7):2017–2033. doi: 10.1093/nar/11.7.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf B., Hsia Y. E., Sweetman L., Gravel R., Harris D. J., Nyhan W. L. Propionic acidemia: a clinical update. J Pediatr. 1981 Dec;99(6):835–846. doi: 10.1016/s0022-3476(81)80004-2. [DOI] [PubMed] [Google Scholar]
- Wolf B., Paulsen E. P., Hsia Y. E. Asymptomatic propionyl CoA carboxylase deficiency in a 13-year-old girl. J Pediatr. 1979 Oct;95(4):563–565. doi: 10.1016/s0022-3476(79)80768-4. [DOI] [PubMed] [Google Scholar]
- Wolf B., Rosenberg L. E. Heterozygote expression in propionyl coenzyme A carboxylase deficiency. Differences between major complementation groups. J Clin Invest. 1978 Nov;62(5):931–936. doi: 10.1172/JCI109221. [DOI] [PMC free article] [PubMed] [Google Scholar]