

Abstract
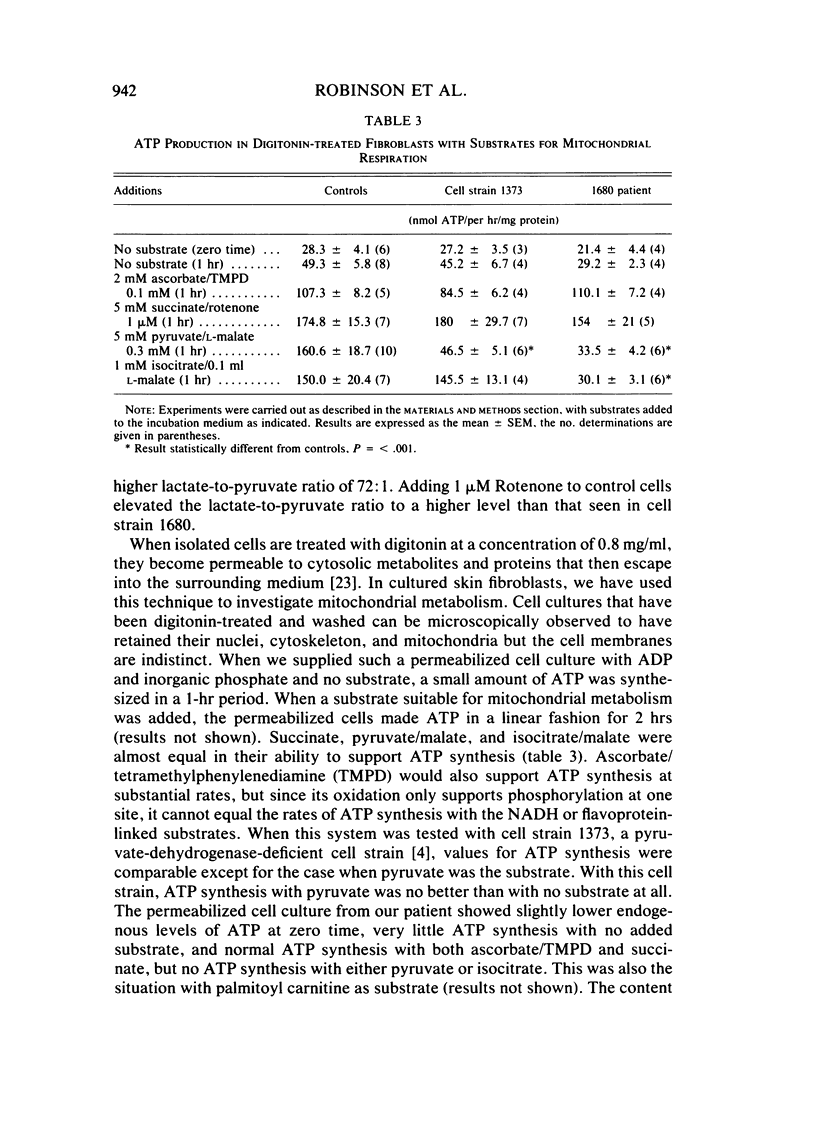
A small-for-gestational-age female infant born at term developed severe lactic acidosis and died on day 13 of life. Two previous sibs had also died of overwhelming lactic acidosis in the neonatal period. The lactate-to-pyruvate and 3-hydroxybutyrate-to-acetoacetate ratios were elevated at 136 and 42 to one, respectively. The activities of the pyruvate dehydrogenase complex and pyruvate carboxylase in cultured skin fibroblasts were normal but a defect in respiration was indicated by the low rates of conversion of 1-[14C]pyruvate, glutamate, and lactate to 14CO2 in these cells. Skin fibroblast cultures also displayed an elevated lactate-to-pyruvate ratio (72:1) when incubated with glucose as substrate compared to control cell cultures (20:1). When mitochondrial preparations of skin fibroblasts (prepared by digitonin extraction) were tested for their ability to synthesize ATP from a variety of substrates, it was found that those of the patient made adequate amounts of ATP with either succinate or ascorbate/tetramethyl-phenylenediamine as substrate but not with the NAD-linked substrates pyruvate, isocitrate, and palmitoyl carnitine. We propose that this is indicative of a defect in the respiratory chain between NADH and coenzyme Q, for the first time demonstrable in cultured skin fibroblasts.
Full text
PDF







Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Anderson S., Bankier A. T., Barrell B. G., de Bruijn M. H., Coulson A. R., Drouin J., Eperon I. C., Nierlich D. P., Roe B. A., Sanger F. Sequence and organization of the human mitochondrial genome. Nature. 1981 Apr 9;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Atkin B. M., Utter M. F., Weinberg M. B. Pyruvate carboxylase and phosphoenolpyruvate carboxykinase activity in leukocytes and fibroblasts from a patient with pyruvate carboxylase deficiency. Pediatr Res. 1979 Jan;13(1):38–43. doi: 10.1203/00006450-197901000-00009. [DOI] [PubMed] [Google Scholar]
- Ballard F. J., Hanson R. W. Phosphoenolpyruvate carboxykinase and pyruvate carboxylase in developing rat liver. Biochem J. 1967 Sep;104(3):866–871. doi: 10.1042/bj1040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. B., Hayes D. J., Byrne E., Morgan-Hughes J. A. Mitochondrial myopathies: defects in mitochondrial metabolism in human skeletal muscle. Biochem Soc Trans. 1983 Dec;11(6):626–627. doi: 10.1042/bst0110626. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Mendell J. R., Sahenk Z., Bachman D., Scarpa A., Scofield R. M., Reiner C. Fatal infantile mitochondrial myopathy and renal dysfunction due to cytochrome-c-oxidase deficiency. Neurology. 1980 Aug;30(8):795–804. doi: 10.1212/wnl.30.8.795. [DOI] [PubMed] [Google Scholar]
- Heiman-Patterson T. D., Bonilla E., DiMauro S., Foreman J., Schotland D. L. Cytochrome-c-oxidase deficiency in a floppy infant. Neurology. 1982 Aug;32(8):898–901. doi: 10.1212/wnl.32.8.898. [DOI] [PubMed] [Google Scholar]
- Heron C., Smith S., Ragan C. I. An analysis of the polypeptide composition of bovine heart mitochondrial NADH-ubiquinone oxidoreductase by two-dimensional polyacrylamide-gel electrophoresis. Biochem J. 1979 Aug 1;181(2):435–443. doi: 10.1042/bj1810435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland K., Leonard J. V. Revised assays for the investigation of congenital lactic acidosis using 14C keto acids, eliminating problems associated with spontaneous decarboxylation. Clin Chim Acta. 1983 Sep 30;133(2):177–187. doi: 10.1016/0009-8981(83)90403-5. [DOI] [PubMed] [Google Scholar]
- Kuźela S., Wielburski A., Nelson B. D. Translation of mitochondrial proteins in digitonin-treated rat hepatocytes. FEBS Lett. 1981 Nov 30;135(1):89–92. doi: 10.1016/0014-5793(81)80950-7. [DOI] [PubMed] [Google Scholar]
- McKay N. D., Robinson B., Brodie R., Rooke-Allen N. Glucose transport and metabolism in cultured human skin fibroblasts. Biochim Biophys Acta. 1983 Apr 5;762(2):198–204. doi: 10.1016/0167-4889(83)90071-x. [DOI] [PubMed] [Google Scholar]
- Miyabayashi S., Narisawa K., Tada K., Sakai K., Kobayashi K., Kobayashi Y. Two siblings with cytochrome c oxidase deficiency. J Inherit Metab Dis. 1983;6(3):121–122. doi: 10.1007/BF01800742. [DOI] [PubMed] [Google Scholar]
- Moreadith R. W., Batshaw M. L., Ohnishi T., Kerr D., Knox B., Jackson D., Hruban R., Olson J., Reynafarje B., Lehninger A. L. Deficiency of the iron-sulfur clusters of mitochondrial reduced nicotinamide-adenine dinucleotide-ubiquinone oxidoreductase (complex I) in an infant with congenital lactic acidosis. J Clin Invest. 1984 Sep;74(3):685–697. doi: 10.1172/JCI111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Hughes J. A., Darveniza P., Kahn S. N., Landon D. N., Sherratt R. M., Land J. M., Clark J. B. A mitochondrial myopathy characterized by a deficiency in reducible cytochrome b. Brain. 1977 Dec;100(4):617–640. doi: 10.1093/brain/100.4.617. [DOI] [PubMed] [Google Scholar]
- Morgan-Hughes J. A., Darveniza P., Landon D. N., Land J. M., Clark J. B. A mitochondrial myopathy with a deficiency of respiratory chain NADH-CoQ reductase activity. J Neurol Sci. 1979 Sep;43(1):27–46. doi: 10.1016/0022-510x(79)90071-6. [DOI] [PubMed] [Google Scholar]
- Robinson B. H. Inborn errors of pyruvate metabolism. Biochem Soc Trans. 1983 Dec;11(6):623–626. doi: 10.1042/bst0110623. [DOI] [PubMed] [Google Scholar]
- Robinson B. H., Oei J., Sherwood W. G., Applegarth D., Wong L., Haworth J., Goodyer P., Casey R., Zaleski L. A. The molecular basis for the two different clinical presentations of classical pyruvate carboxylase deficiency. Am J Hum Genet. 1984 Mar;36(2):283–294. [PMC free article] [PubMed] [Google Scholar]
- Robinson B. H., Sherwood W. G. Lactic acidaemia. J Inherit Metab Dis. 1984;7 (Suppl 1):69–73. doi: 10.1007/BF03047378. [DOI] [PubMed] [Google Scholar]
- Robinson B. H., Taylor J., Francois B., Beaudet A. L., Peterson D. F. Lactic acidosis, neurological deterioration and compromised cellular pyruvate oxidation due to a defect in the reoxidation of cytoplasmically generated NADH. Eur J Pediatr. 1983 Apr;140(2):98–101. doi: 10.1007/BF00441651. [DOI] [PubMed] [Google Scholar]
- Robinson B. H., Taylor J., Sherwood W. G. The genetic heterogeneity of lactic acidosis: occurrence of recognizable inborn errors of metabolism in pediatric population with lactic acidosis. Pediatr Res. 1980 Aug;14(8):956–962. doi: 10.1203/00006450-198008000-00013. [DOI] [PubMed] [Google Scholar]
- Rosamond J. The molecular biology of the mitochondrion. Biochem J. 1982 Jan 15;202(1):1–8. doi: 10.1042/bj2020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu K. F., Hu C. W., Utter M. F. Pyruvate dehydrogenase complex activity in normal and deficient fibroblasts. J Clin Invest. 1981 May;67(5):1463–1471. doi: 10.1172/JCI110176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro A. J., Moore C. L., Prineas J. W., Strasberg P. M., Rapin I. A cytochrome-related inherited disorder of the nervous system and muscle. Arch Neurol. 1970 Aug;23(2):103–112. doi: 10.1001/archneur.1970.00480260009002. [DOI] [PubMed] [Google Scholar]
- Van Biervliet J. P., Bruinvis L., Ketting D., De Bree P. K., Van der Heiden C., Wadman S. K. Hereditary mitochondrial myopathy with lactic acidemia, a De Toni-Fanconi-Debré syndrome, and a defective respiratory chain in voluntary striated muscles. Pediatr Res. 1977 Oct;11(10 Pt 2):1088–1093. doi: 10.1203/00006450-197711100-00005. [DOI] [PubMed] [Google Scholar]
- Willems J. L., Monnens L. A., Trijbels J. M., Veerkamp J. H., Meyer A. E., van Dam K., van Haelst U. Leigh's encephalomyelopathy in a patient with cytochrome c oxidase deficiency in muscle tissue. Pediatrics. 1977 Dec;60(6):850–857. [PubMed] [Google Scholar]