

Abstract
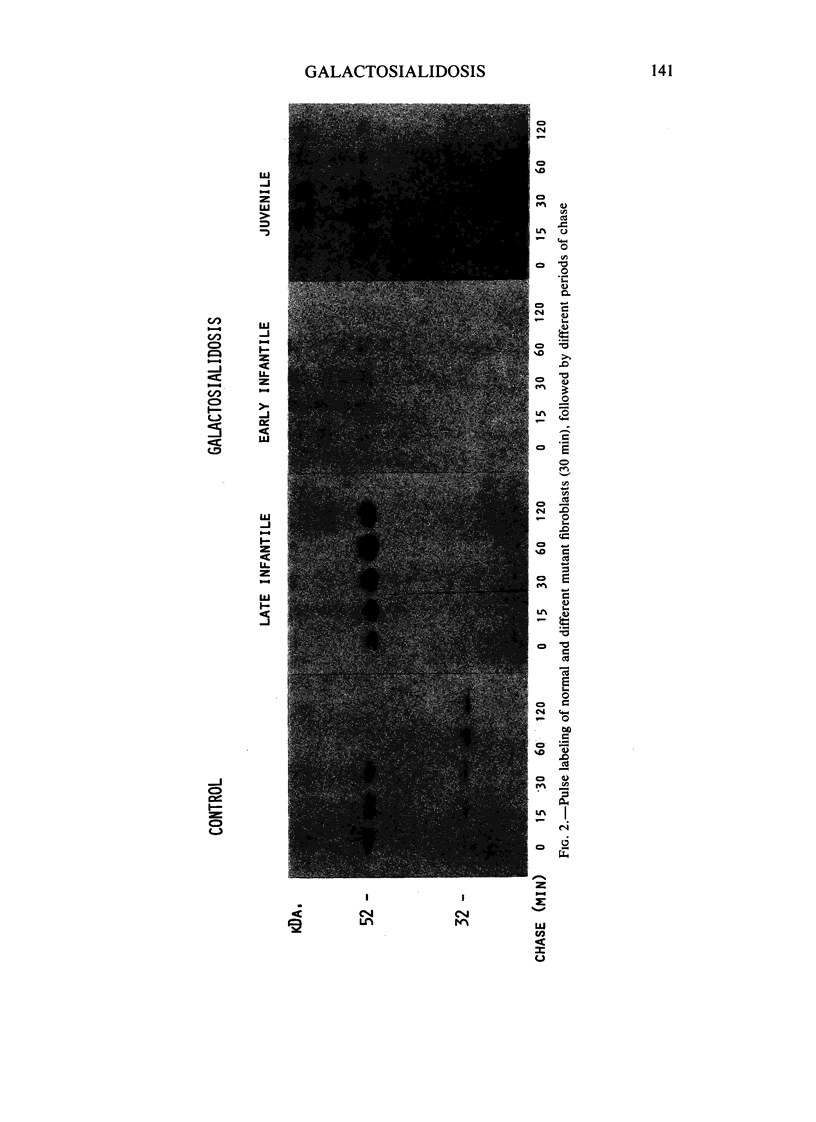
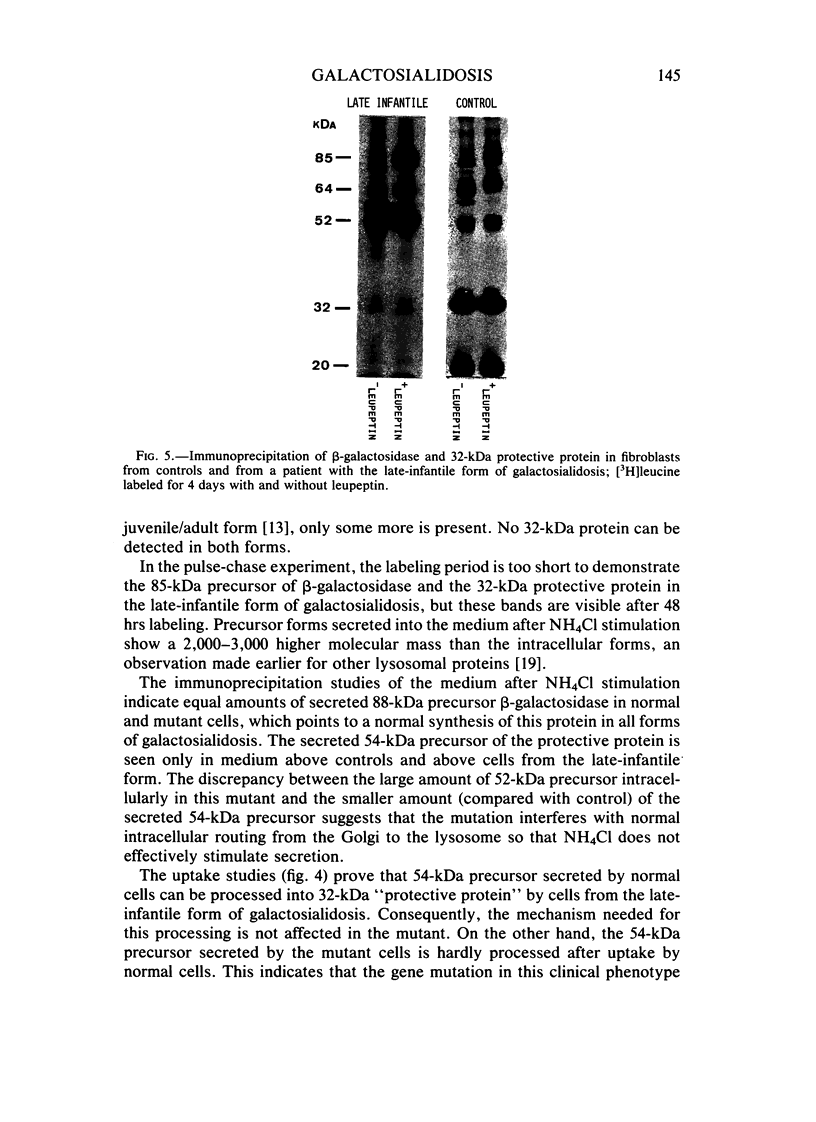
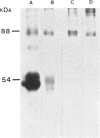
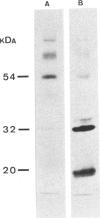
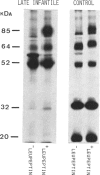
The lysosomal storage disorder galactosialidosis has been recognized as a distinct genetic and biochemical entity, associated with a combined beta-galactosidase and neuraminidase deficiency that is due to the lack of a 32-kilodalton (kDa) glycoprotein. The molecular basis of different clinical variants of galactosialidosis has been investigated. In the early-infantile form, the synthesis of the 52-kDa precursor of the 32-kDa "protective protein" is markedly reduced and the absence of the latter protein explains the severe neuraminidase deficiency. In the juvenile-adult form, there is relatively more 52-kDa precursor but no 32-kDa protein can be detected. Cells from the late-infantile form have in comparison with controls, besides a small amount of the 32-kDa glycoprotein, an accumulation of the 52-kDa precursor. Apparently, this protein is genetically altered in such a way that its further processing is impaired. Furthermore, in this mutant, the residual neuraminidase activity is stimulated four- to sixfold upon leupeptin treatment together with an increase of the 32-kDa glycoprotein.
Full text
PDF









Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Conzelmann E., Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6(1):58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- D'Azzo A., Hoogeveen A., Reuser A. J., Robinson D., Galjaard H. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci U S A. 1982 Aug;79(15):4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel R. A., Lowden J. A., Callahan J. W., Wolfe L. S., Ng Yin Kin N. M. Infantile sialidosis: a phenocopy of type 1 GM1 gangliosidosis distinguished by genetic complementation and urinary oligosaccharides. Am J Hum Genet. 1979 Nov;31(6):669–679. [PMC free article] [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J Biol Chem. 1980 May 25;255(10):4946–4950. [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1980 May 25;255(10):4937–4945. [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1980 May 25;255(10):4937–4945. [PubMed] [Google Scholar]
- Kleijer W. J., Hoogeveen A., Verheijen F. W., Niermeijer M. F., Galjaard H., O'Brien J. S., Warner T. G. Prenatal diagnosis of sialidosis with combined neuraminidase and beta-galactosidase deficiency. Clin Genet. 1979 Jul;16(1):60–61. doi: 10.1111/j.1399-0004.1979.tb00851.x. [DOI] [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Loonen M. C., Lugt L., Franke C. L. Letter: Angiokeratoma corporis diffusum and lysosomal enzyme deficiency. Lancet. 1974 Sep 28;2(7883):785–785. doi: 10.1016/s0140-6736(74)90984-2. [DOI] [PubMed] [Google Scholar]
- Loonen M. C., Reuser A. J., Visser P., Arts W. F. Combined sialidase (neuraminidase) and beta-galactosidase deficiency. Clinical, morphological and enzymological observations in a patient. Clin Genet. 1984 Aug;26(2):139–149. doi: 10.1111/j.1399-0004.1984.tb00804.x. [DOI] [PubMed] [Google Scholar]
- Lowden J. A., O'Brien J. S. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979 Jan;31(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- Norden A. G., Tennant L. L., O'Brien J. S. GM1 ganglioside beta-galactosidase. A. Purification and studies of the enzyme from human liver. J Biol Chem. 1974 Dec 25;249(24):7969–7976. [PubMed] [Google Scholar]
- Penefsky H. S. Reversible binding of Pi by beef heart mitochondrial adenosine triphosphatase. J Biol Chem. 1977 May 10;252(9):2891–2899. [PubMed] [Google Scholar]
- Pinsky L., Miller J., Shanfield B., Watters G., Wolfe L. S. GM1 gangliosidosis in skin fibroblast culture: enzymatic differences between types 1 and 2 and observations on a third variant. Am J Hum Genet. 1974 Sep;26(5):563–577. [PMC free article] [PubMed] [Google Scholar]
- Strisciuglio P., Creek K. E., Sly W. S. Complementation, cross correction, and drug correction studies of combined beta-galactosidase neuraminidase deficiency in human fibroblasts. Pediatr Res. 1984 Feb;18(2):167–171. doi: 10.1203/00006450-198402000-00011. [DOI] [PubMed] [Google Scholar]
- Verheijen F. W., Palmeri S., Hoogeveen A. T., Galjaard H. Human placental neuraminidase. Activation, stabilization and association with beta-galactosidase and its protective protein. Eur J Biochem. 1985 Jun 3;149(2):315–321. doi: 10.1111/j.1432-1033.1985.tb08928.x. [DOI] [PubMed] [Google Scholar]
- Verheijen F., Brossmer R., Galjaard H. Purification of acid beta-galactosidase and acid neuraminidase from bovine testis: evidence for an enzyme complex. Biochem Biophys Res Commun. 1982 Sep 30;108(2):868–875. doi: 10.1016/0006-291x(82)90911-1. [DOI] [PubMed] [Google Scholar]
- Wenger D. A., Tarby T. J., Wharton C. Macular cherry-red spots and myoclonus with dementia: coexistent neuraminidase and beta-galactosidase deficiencies. Biochem Biophys Res Commun. 1978 May 30;82(2):589–595. doi: 10.1016/0006-291x(78)90915-4. [DOI] [PubMed] [Google Scholar]