

Abstract
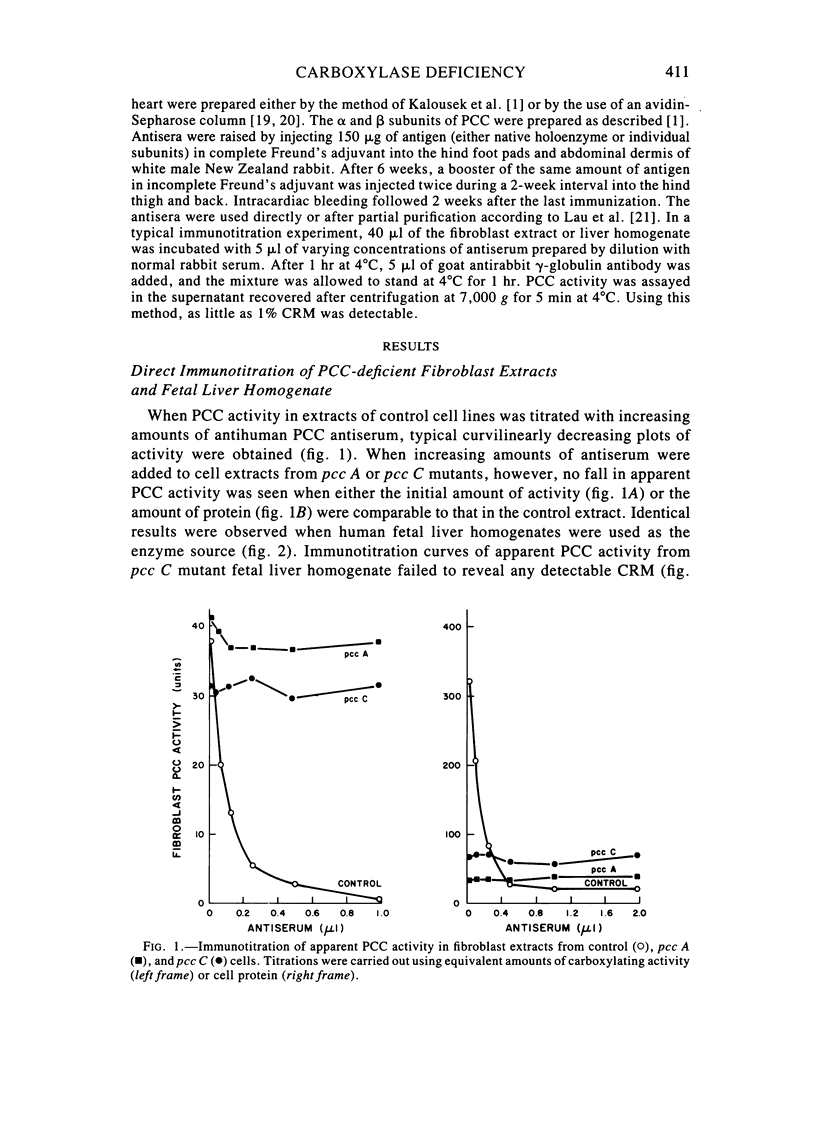
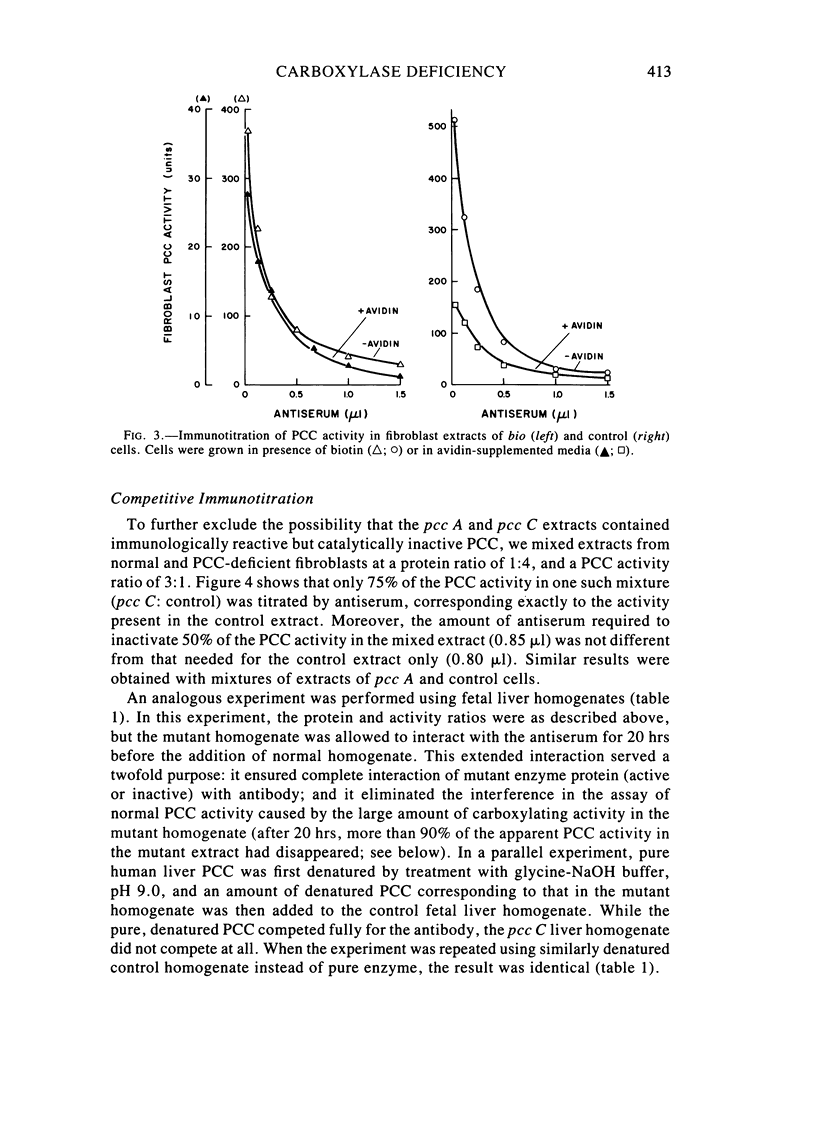
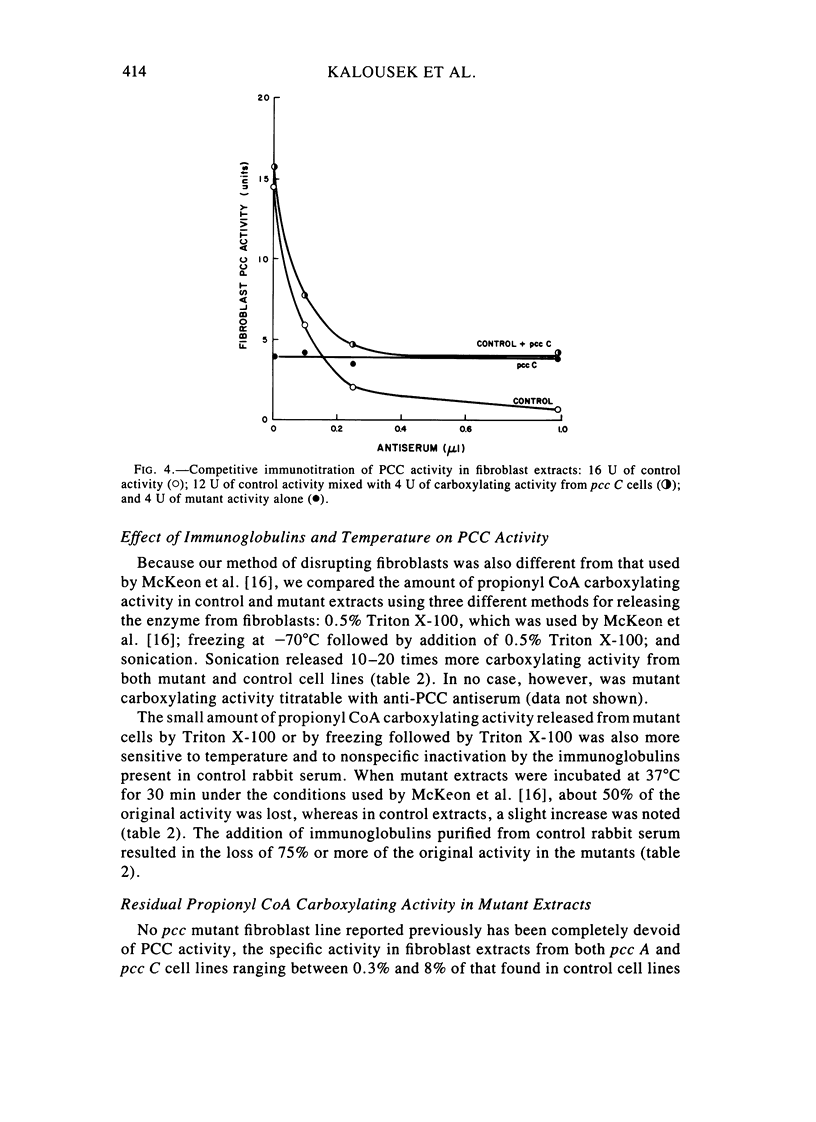
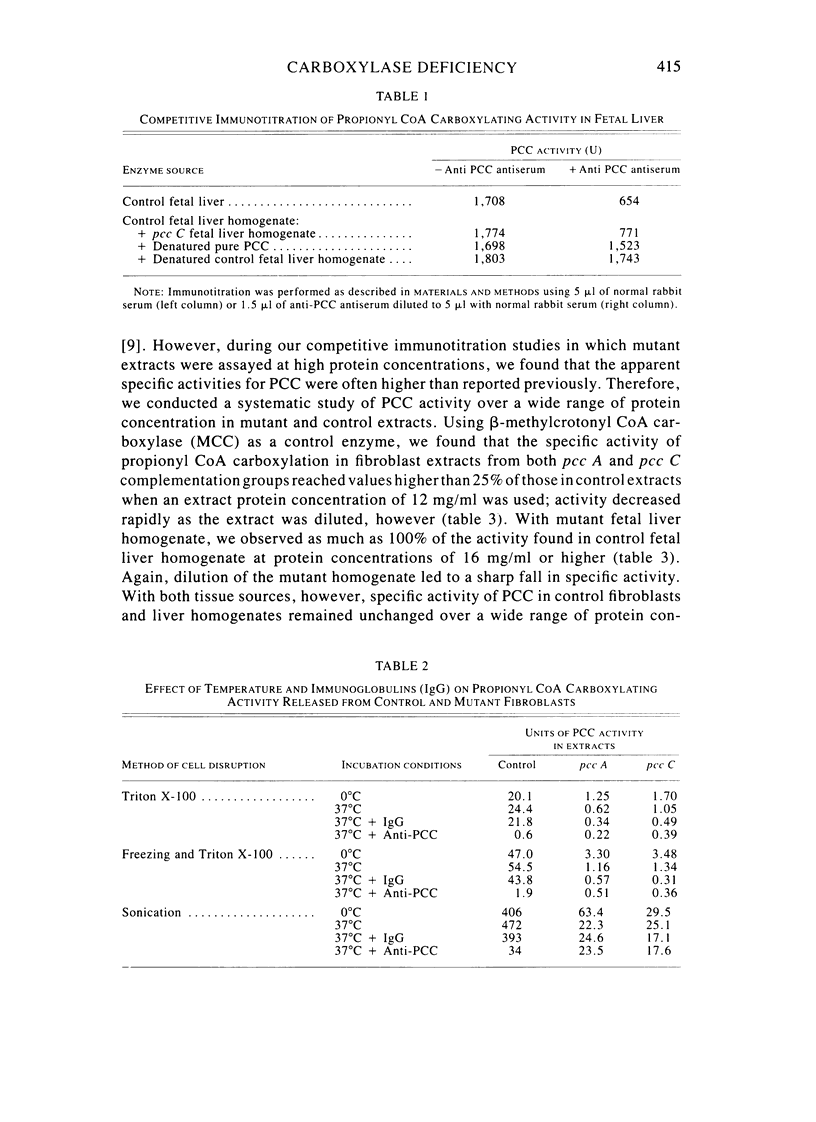
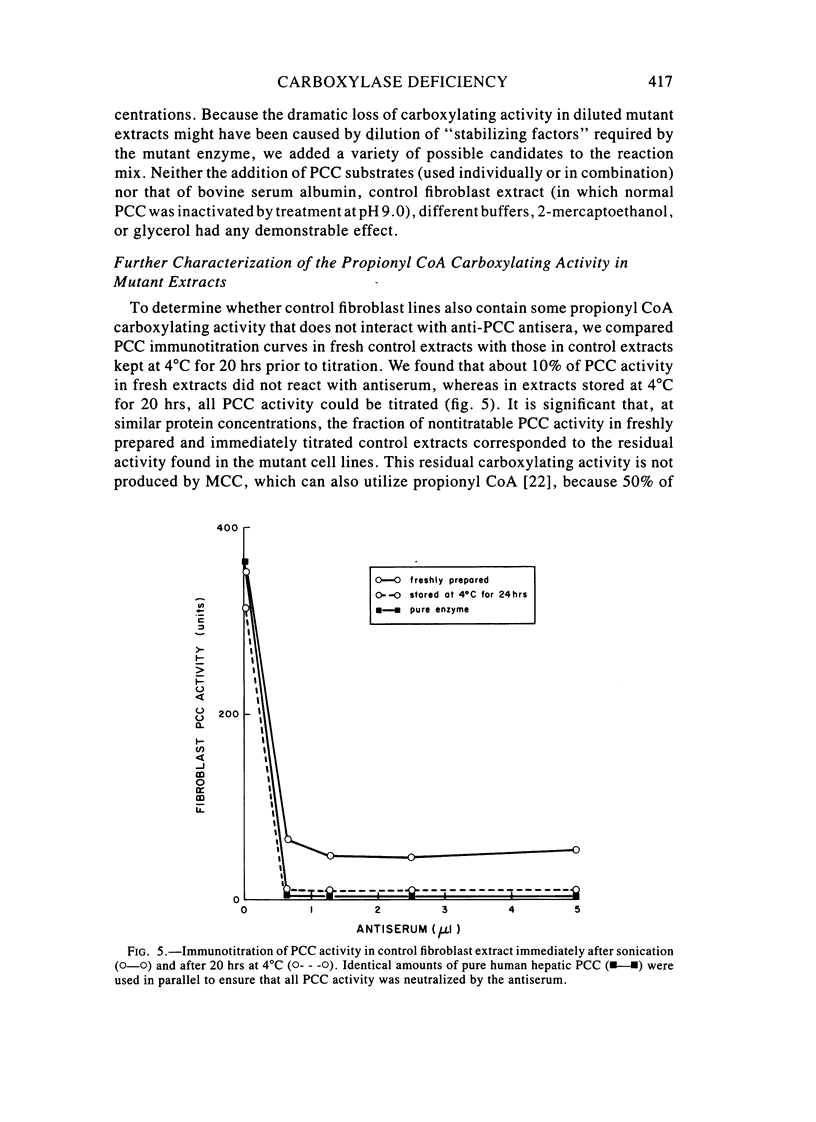
Fibroblast extracts and fetal liver homogenates from patients with propionic acidemia due to inherited deficiency of propionyl CoA carboxylase (PCC) were analyzed for the presence of immunologically cross-reactive PCC protein. Using several rabbit antisera raised against homogeneous human liver PCC, homogeneous pig heart PCC, or the individual non-identical subunits of the human liver enzyme, we found no detectable cross-reacting material by direct or competitive immunotitration in several cell lines from patients in either major complementation group (pcc A; pcc C) with isolated PCC deficiency. In contrast, cells of a patient from the bio complementation group contained normal amounts of immunoreactive PCC. Further analysis of the pcc A and pcc C mutants revealed that their residual propionyl CoA carboxylating activity varied greatly depending on the concentration of extract or homogenate protein used in the PCC assay. When propionyl CoA carboxylation was assayed at high protein concentration in a fetal liver homogenate from a pcc C patient, the apparent PCC activity was comparable to that found in normal human fetal liver. Significantly, the specific activity in the mutant, but not in the control, extract declined steeply as protein concentration was lowered, and this loss could not be prevented by adding PCC substrates, bovine serum albumin, glycerol, or 2-mercaptoethanol. Moreover, detailed analyses of immunotitration curves of control fibroblasts extracts showed that fresh extracts contained an amount of nonimmunotitratable carboxylating activity corresponding to the residual activity present in fresh extracts of mutant cell lines. We conclude that the residual propionyl CoA carboxylating activity found in isolated PCC deficiency represents another carboxylase that can utilize propionyl CoA as a substrate rather than a mutant form of PCC with markedly different immunochemical and physicochemical properties.
Full text
PDF






Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Ando T., Rasmussen K., Nyhan W. L., Donnell G. N., Barnes N. D. Propionic acidemia in patients with ketotic hyperglycinemia. J Pediatr. 1971 May;78(5):827–832. doi: 10.1016/s0022-3476(71)80354-2. [DOI] [PubMed] [Google Scholar]
- Bartlett K., Gompertz D. Combined carboxylase defect: biotin-responsiveness in cultured fibroblasts. Lancet. 1976 Oct 9;2(7989):804–804. doi: 10.1016/s0140-6736(76)90640-1. [DOI] [PubMed] [Google Scholar]
- Burri B. J., Sweetman L., Nyhan W. L. Mutant holocarboxylase synthetase: evidence for the enzyme defect in early infantile biotin-responsive multiple carboxylase deficiency. J Clin Invest. 1981 Dec;68(6):1491–1495. doi: 10.1172/JCI110402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman G. L., Wolf B. Deficient acetyl CoA carboxylase activity in multiple carboxylase deficiency. Clin Chim Acta. 1981 Apr 9;111(2-3):147–151. doi: 10.1016/0009-8981(81)90181-9. [DOI] [PubMed] [Google Scholar]
- GREEN N. M. AVIDIN. 1. THE USE OF (14-C)BIOTIN FOR KINETIC STUDIES AND FOR ASSAY. Biochem J. 1963 Dec;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompertz D., Storrs C. N., Bau D. C., Peters T. J., Hughes E. A. Localisation of enzymic defect in propionicacidaemia. Lancet. 1970 May 30;1(7657):1140–1143. doi: 10.1016/s0140-6736(70)91216-x. [DOI] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Mahuran D., Kronis A. Purification of human liver propionyl-CoA carboxylase by carbon tetrachloride extraction and monomeric avidin affinity chromatography. Arch Biochem Biophys. 1980 May;201(2):669–673. doi: 10.1016/0003-9861(80)90557-3. [DOI] [PubMed] [Google Scholar]
- Gravel R. A., Lam K. F., Scully K. J., Hsia Y. Genetic complementation of propionyl-CoA carboxylase deficiency in cultured human fibroblasts. Am J Hum Genet. 1977 Jul;29(4):378–388. [PMC free article] [PubMed] [Google Scholar]
- Hsia Y. E., Scully K. J. Propionic acidemia: diagnosis by enzyme assay in frozen leukocytes. J Pediatr. 1973 Oct;83(4):625–628. doi: 10.1016/s0022-3476(73)80226-4. [DOI] [PubMed] [Google Scholar]
- Hsia Y. E., Scully K. J., Rosenberg L. E. Defective propionate carboxylation in ketotic hyperglycinaemia. Lancet. 1969 Apr 12;1(7598):757–758. doi: 10.1016/s0140-6736(69)91757-7. [DOI] [PubMed] [Google Scholar]
- Hsia Y. E., Scully K. J., Rosenberg L. E. Human propionyl CoA carboxylase: some properties of the partially purified enzyme in fibroblasts from controls and patients with propionic acidemia. Pediatr Res. 1979 Jun;13(6):746–751. doi: 10.1203/00006450-197906000-00005. [DOI] [PubMed] [Google Scholar]
- Hsia Y. E., Scully K. J., Rosenberg L. E. Inherited propionyl-Coa carboxylase deficiency in "ketotic hyperglycinemia". J Clin Invest. 1971 Jan;50(1):127–130. doi: 10.1172/JCI106466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalousek F., Darigo M. D., Rosenberg L. E. Isolation and characterization of propionyl-CoA carboxylase from normal human liver. Evidence for a protomeric tetramer of nonidentical subunits. J Biol Chem. 1980 Jan 10;255(1):60–65. [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Lau E. P., Cochran B. C., Munson L., Fall R. R. Bovine kidney 3-methylcrotonyl-CoA and propionyl-CoA carboxylases: each enzyme contains nonidentical subunits. Proc Natl Acad Sci U S A. 1979 Jan;76(1):214–218. doi: 10.1073/pnas.76.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon C., Eanes R. Z., Fall R. R., Tasset D. M., Wolf B. Immunological studies of propionyl CoA carboxylase in livers and fibroblasts of patients with propionic acidemia. Clin Chim Acta. 1980 Feb 28;101(2-3):217–233. doi: 10.1016/0009-8981(80)90246-6. [DOI] [PubMed] [Google Scholar]
- Saunders M. E., Sherwood W. G., Duthie M., Surh L., Gravel R. A. Evidence for a defect of holocarboxylase synthetase activity in cultured lymphoblasts from a patient with biotin-responsive multiple carboxylase deficiency. Am J Hum Genet. 1982 Jul;34(4):590–601. [PMC free article] [PubMed] [Google Scholar]
- Saunders M., Sweetman L., Robinson B., Roth K., Cohn R., Gravel R. A. Biotin-response organicaciduria. Multiple carboxylase defects and complementation studies with propionicacidemia in cultured fibroblasts. J Clin Invest. 1979 Dec;64(6):1695–1702. doi: 10.1172/JCI109632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyler W., Sweetman L., Maggio D. C., Nyhan W. L. Deficiency of propionyl-Co A carboxylase and methylcrotonyl-Co A carboxylase in a patient with methylcrotonylglycinuria. Clin Chim Acta. 1977 May 2;76(3):321–328. doi: 10.1016/0009-8981(77)90158-9. [DOI] [PubMed] [Google Scholar]
- Wolf B., Hsia Y. E., Rosenberg L. E. Biochemical differences between mutant propionyl-CoA carboxylases from two complementation groups. Am J Hum Genet. 1978 Sep;30(5):455–464. [PMC free article] [PubMed] [Google Scholar]