

Abstract
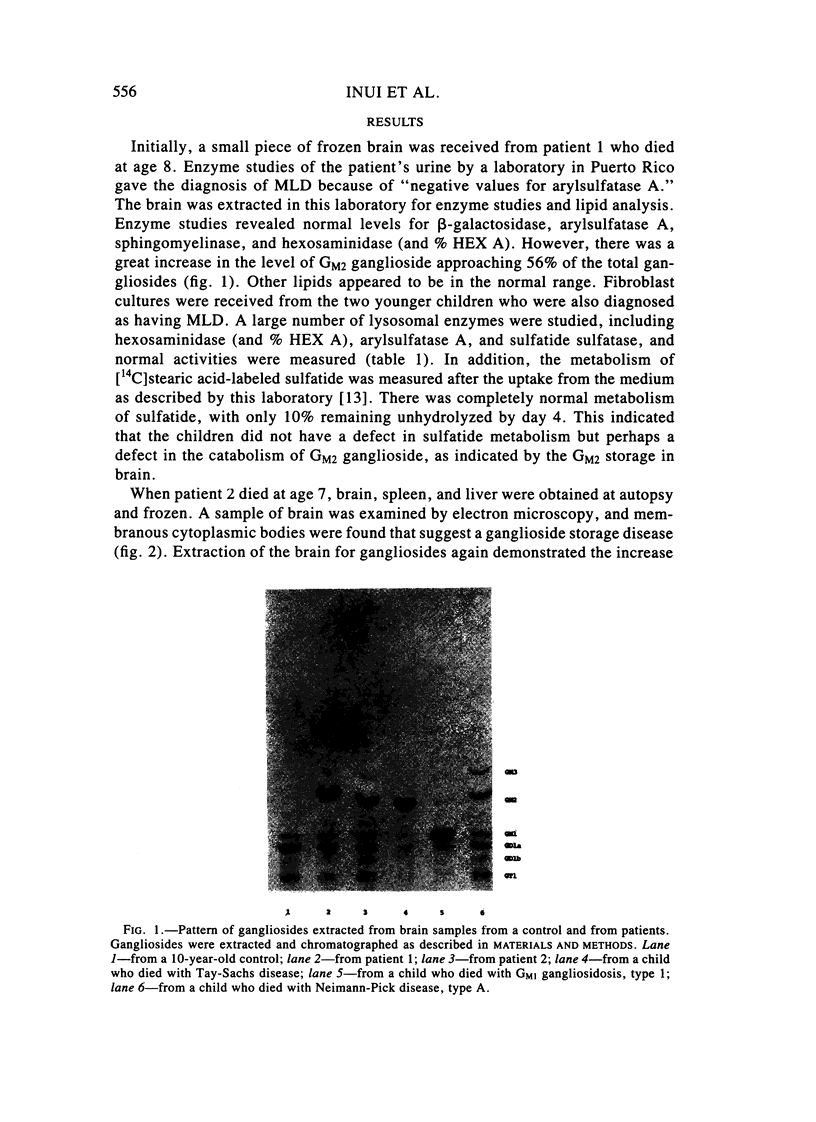
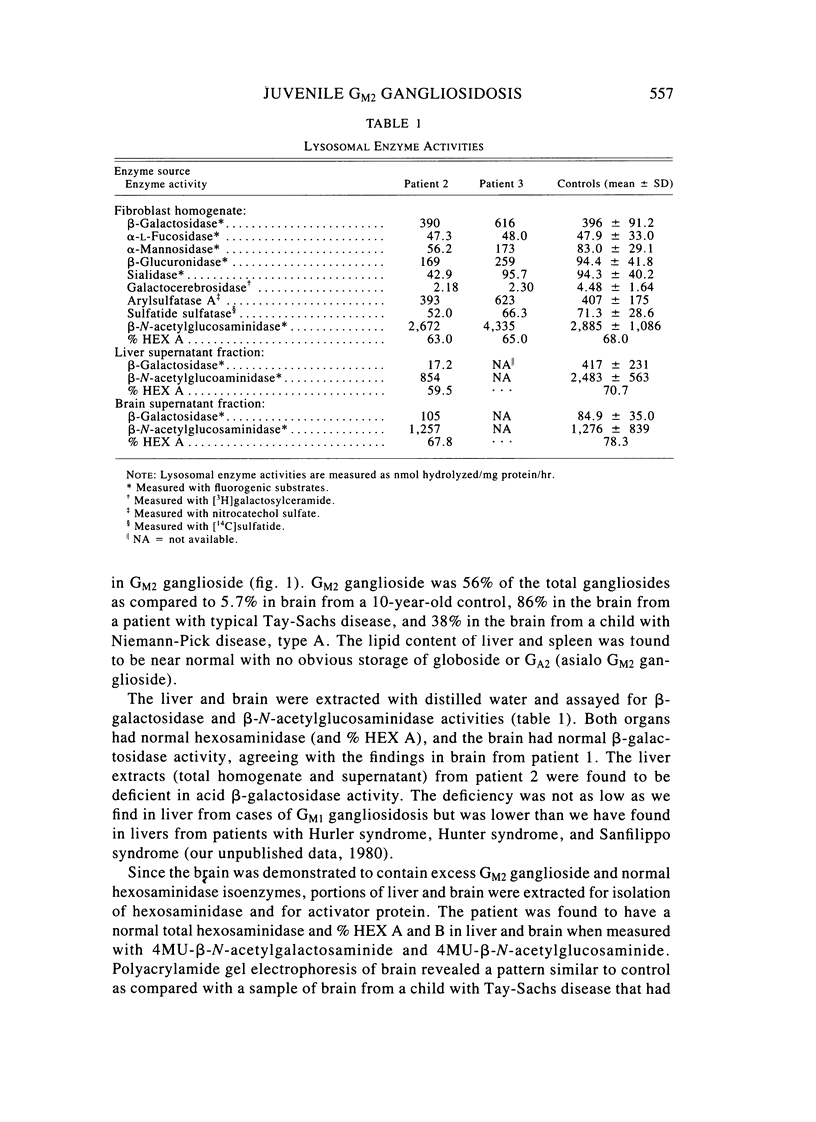
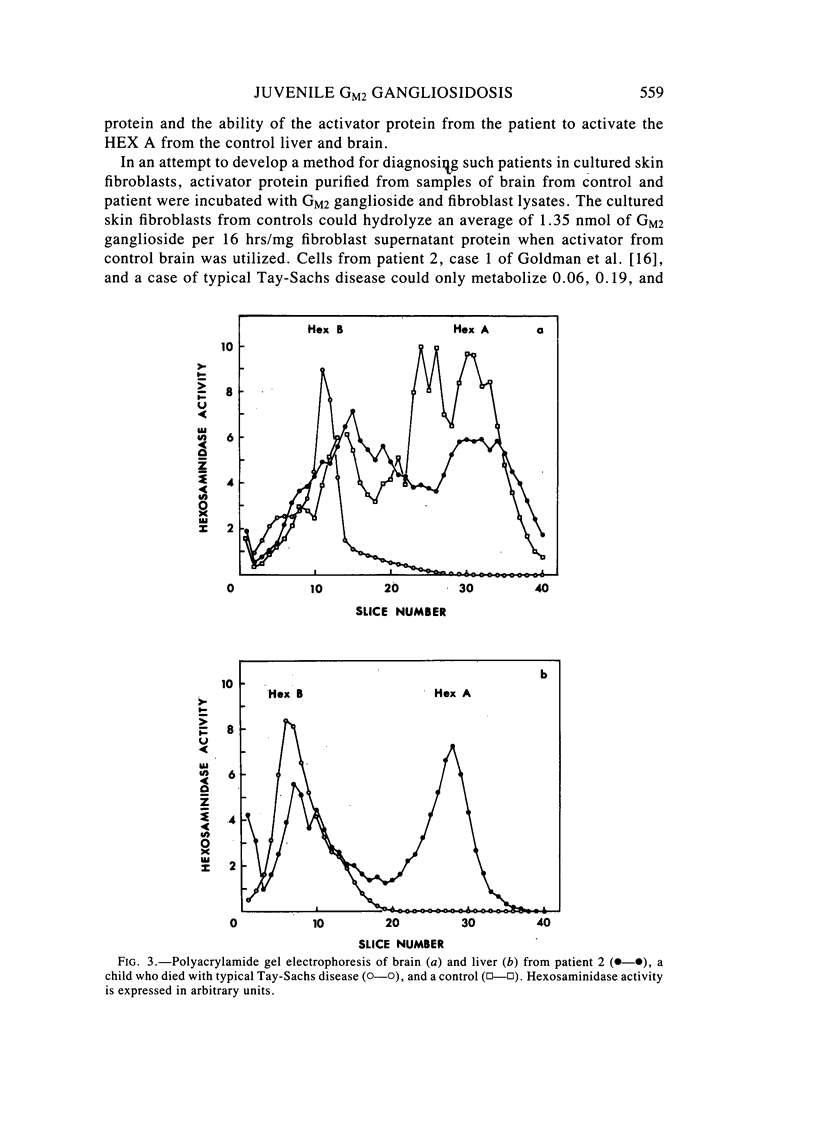
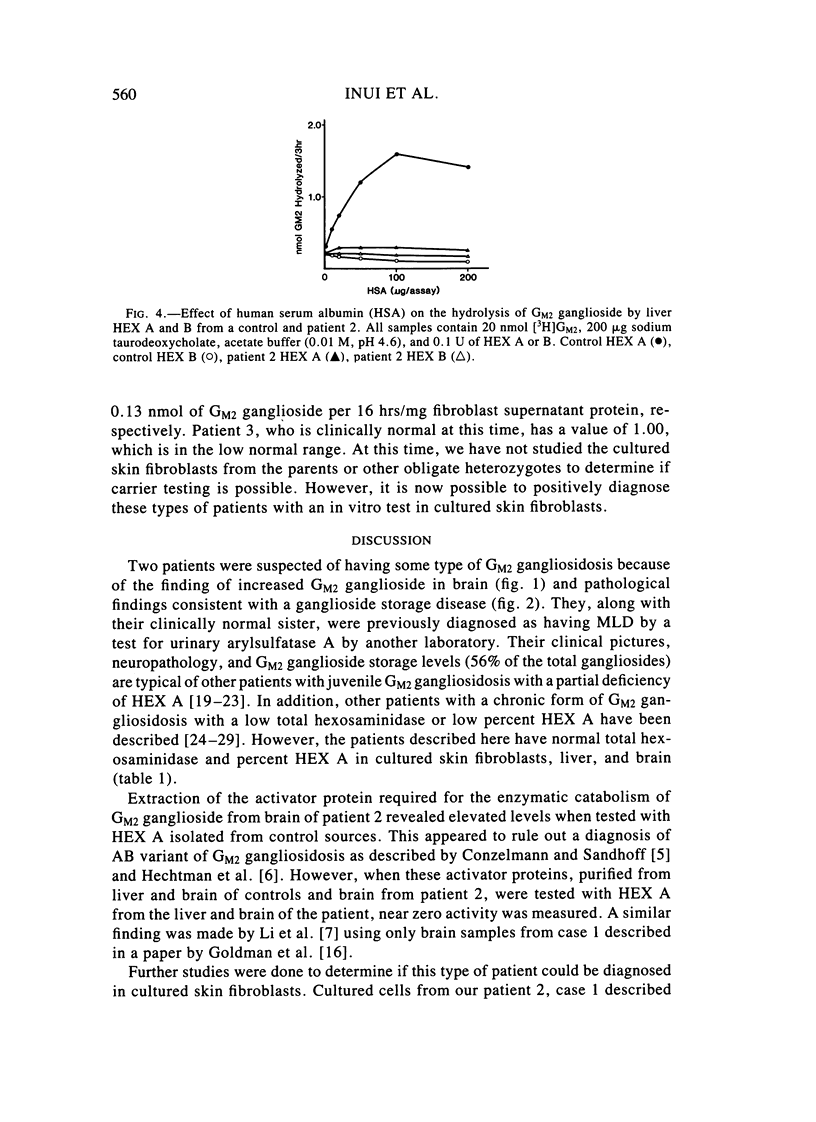
Two sibling from a consanguineous Puerto Rican marriage were found to have a juvenile-onset type of lipidosis first noted at age 2 1/2 by expressing difficulties with motor function and developmental delay. They continued to deteriorate, showing muscle atrophy, spasticity, and loss of speech, and death occurred at ages 7 and 8. Examination of the brains from these patients revealed that the concentration of GM2 ganglioside was about 56% of the total gangliosides. Hexosaminidase and percent hexosaminidase A (HEX A) and other lysosomal enzymes were normal in cultured skin fibroblasts, liver, and brain. The concentration of the activator protein required for the enzymatic hydrolysis of GM2 ganglioside was in high normal levels in the brain of the patient available. However, the HEX A from the patient's brain and liver as well as from skin fibroblast lysates could not be activated to hydrolyze GM2 ganglioside by the activator protein from a control or himself. The HEX A from a control could be activated by the activator protein from controls or this patient. These patients appear to have a defect in HEX A, which does not affect it heat stability, electrophoretic migration, and activity toward fluorogenic substrates, but may affect the binding of the activator protein required for GM2 ganglioside hydrolysis. We propose to call these patients the AMB variant of GM2 gangliosidosis to denote the mutation in HEX A but with normal levels of HEX A and B with synthetic substrates. This is to distinguish these patients from those missing the activator protein and normal HEX A and B levels.
Full text
PDF





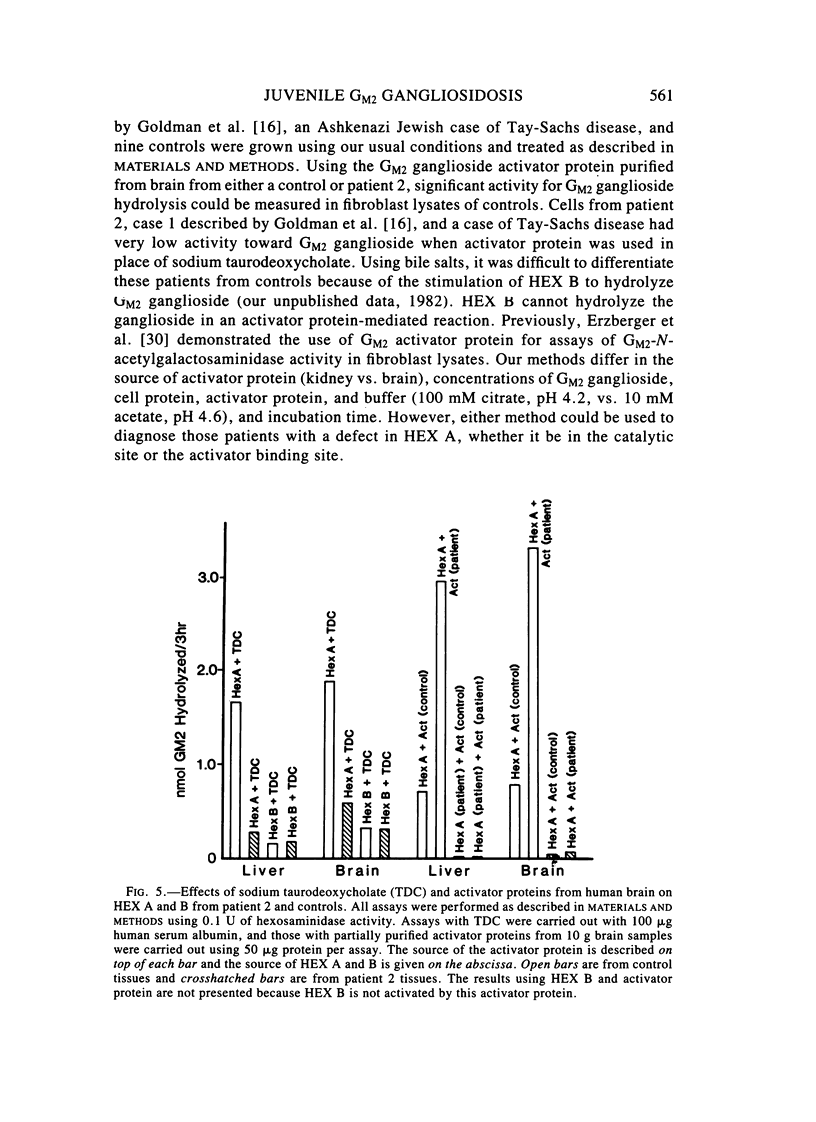



Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Brett E. M., Ellis R. B., Haas L., Ikonne J. U., Lake B. D., Patrick A. D., Stephens R. Late onset GM2-gangliosidosis. Clinical, pathological, and biochemical studies on 8 patients. Arch Dis Child. 1973 Oct;48(10):775–785. doi: 10.1136/adc.48.10.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester M. A., Hultberg B., Liedholm H., Ockerman P. A. A new N-acetyl-beta-D-hexosaminidase disease with late onset of progressive neurological symptoms. Hum Hered. 1979;29(2):124–128. doi: 10.1159/000153028. [DOI] [PubMed] [Google Scholar]
- Conzelmann E., Sandhoff K. AB variant of infantile GM2 gangliosidosis: deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2. Proc Natl Acad Sci U S A. 1978 Aug;75(8):3979–3983. doi: 10.1073/pnas.75.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann E., Sandhoff K., Nehrkorn H., Geiger B., Arnon R. Purification, biochemical and immunological characterisation of hexosaminidase A from variant AB of infantile GM2 gangliosidosis. Eur J Biochem. 1978 Mar;84(1):27–33. doi: 10.1111/j.1432-1033.1978.tb12137.x. [DOI] [PubMed] [Google Scholar]
- Dubois G., Zalc B., Le Saux F., Baumann N. Stearoyl[1-14C]sulfogalactosylsphingosine ([14C]sulfatide) as substrate for cerebroside sulfatase assay. Anal Biochem. 1980 Mar 1;102(2):313–317. doi: 10.1016/0003-2697(80)90159-1. [DOI] [PubMed] [Google Scholar]
- Erzberger A., Conzelmann E., Sandhoff K. Assay of ganglioside GM2-N-acetyl-beta-D-galactosaminidase activity in human fibroblasts employing the natural activator protein--diagnosis of variant forms of GM2 gangliosidosis. Clin Chim Acta. 1980 Dec 22;108(3):361–368. doi: 10.1016/0009-8981(80)90342-3. [DOI] [PubMed] [Google Scholar]
- Goldman J. E., Yamanaka T., Rapin I., Adachi M., Suzuki K., Suzuki K. The AB-variant of GM2-gangliosidosis. Clinical, biochemical, and pathological studies of two patients. Acta Neuropathol. 1980;52(3):189–202. doi: 10.1007/BF00705807. [DOI] [PubMed] [Google Scholar]
- Grebner E. E., Jackson L. G. Prenatal diagnosis of Tay-Sachs disease: studies on the reliability of hexosaminidase levels in amniotic fluid. Am J Obstet Gynecol. 1979 Jul 1;134(5):547–550. doi: 10.1016/0002-9378(79)90838-x. [DOI] [PubMed] [Google Scholar]
- Hechtman P., Gordon B. A., Ng Ying Kin N. M. Deficiency of the hexosaminidase A activator protein in a case of GM2 gangliosidosis; variant AB. Pediatr Res. 1982 Mar;16(3):217–222. doi: 10.1203/00006450-198203000-00011. [DOI] [PubMed] [Google Scholar]
- Johnson W. G., Chutorian A., Miranda A. A new juvenile hexosaminidase deficiency disease presenting as cerebellar ataxia. Clinical and biochemical studies. Neurology. 1977 Nov;27(11):1012–1018. doi: 10.1212/wnl.27.11.1012. [DOI] [PubMed] [Google Scholar]
- Kudoh T., Sattler M., Malmstrom J., Bitter M. A., Wenger D. A. Metabolism of fatty acid-labeled cerebroside sulfate in cultured cells from controls and metachromatic leukodystrophy patients. Use in the prenatal identification of a false positive fetus. J Lab Clin Med. 1981 Nov;98(5):704–714. [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Li S. C., Hirabayashi Y., Li Y. T. A new variant of type-AB GM2-gangliosidosis. Biochem Biophys Res Commun. 1981 Jul 30;101(2):479–485. doi: 10.1016/0006-291x(81)91285-7. [DOI] [PubMed] [Google Scholar]
- Menkes J. H., O'Brien J. S., Okada S., Grippo J., Andrews J. M., Cancilla P. A. Juvenile GM2 gangliosidosis. Biochemical and ultrastructural studies on a new variant of Tay-Sachs disease. Arch Neurol. 1971 Jul;25(1):14–22. doi: 10.1001/archneur.1971.00490010024003. [DOI] [PubMed] [Google Scholar]
- O'Brien J. S., Norden G. W., Miller A. L., Frost R. G., Kelly T. E. Ganglioside GM2 N-acetyl-beta-D-galactosaminidase and asialo GM2 (GA2) N-acetyl-beta-D-galactosaminidase; studies in human skin fibroblasts. Clin Genet. 1977 Mar;11(3):171–183. doi: 10.1111/j.1399-0004.1977.tb01296.x. [DOI] [PubMed] [Google Scholar]
- Okada S., O'Brien J. S. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science. 1969 Aug 15;165(3894):698–700. doi: 10.1126/science.165.3894.698. [DOI] [PubMed] [Google Scholar]
- Okada S., O'Brien J. S. Tay-Sachs disease: generalized absence of a beta-D-N-acetylhexosaminidase component. Science. 1969 Aug 15;165(3894):698–700. doi: 10.1126/science.165.3894.698. [DOI] [PubMed] [Google Scholar]
- Okada S., Veath M. L., O'Brien J. S. Juvenile GM2 gangliosidosis: partial deficiency of hexosaminidase A. J Pediatr. 1970 Dec;77(6):1063–1065. doi: 10.1016/s0022-3476(70)80096-8. [DOI] [PubMed] [Google Scholar]
- Oonk J. G., van der Helm H. J., Martin J. J. Spinocerebellar degeneration: hexosaminidase A and B deficiency in two adult sisters. Neurology. 1979 Mar;29(3):380–384. doi: 10.1212/wnl.29.3.380. [DOI] [PubMed] [Google Scholar]
- Radin N. S., Hof L., Bradley R. M., Brady R. O. Lactosylceramide galactosidase: comparison with other sphingolipid hydrolases in developing rat brain. Brain Res. 1969 Jul;14(2):497–505. doi: 10.1016/0006-8993(69)90124-3. [DOI] [PubMed] [Google Scholar]
- Rapin I., Suzuki K., Suzuki K., Valsamis M. P. Adult (chronic) GM2 gangliosidosis. Atypical spinocerebellar degeneration in a Jewish sibship. Arch Neurol. 1976 Feb;33(2):120–130. doi: 10.1001/archneur.1976.00500020048008. [DOI] [PubMed] [Google Scholar]
- SVENNERHOLM L. Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim Biophys Acta. 1957 Jun;24(3):604–611. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- Sandhoff K., Christomanou H. Biochemistry and genetics of gangliosidoses. Hum Genet. 1979;50(2):107–143. doi: 10.1007/BF00390234. [DOI] [PubMed] [Google Scholar]
- Sandhoff K., Christomanou H. Biochemistry and genetics of gangliosidoses. Hum Genet. 1979;50(2):107–143. doi: 10.1007/BF00390234. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 1969 Aug;4(4):351–354. doi: 10.1016/0014-5793(69)80274-7. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. Variation of beta-N-acetylhexosaminidase-pattern in Tay-Sachs disease. FEBS Lett. 1969 Aug;4(4):351–354. doi: 10.1016/0014-5793(69)80274-7. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Rapin I., Suzuki Y., Ishii N. Juvenile GM2-gangliosidosis. Clinical variant of Tay-Sachs disease or a new disease. Neurology. 1970 Feb;20(2):190–204. doi: 10.1212/wnl.20.2.190. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Suzuki K. Partial deficiency of hexosaminidase component a in juvenile gm2-gangliosidosis. Neurology. 1970 Sep;20(9):848–851. doi: 10.1212/wnl.20.9.848. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Suzuki K. Specific radioactive labeling of terminal n-acetylgalactosamine of glycosphingolipids by the galactose oxidase-sodium borohydride method. J Lipid Res. 1972 Sep;13(5):687–690. [PubMed] [Google Scholar]
- Warner T. G., O'Brien J. S. Synthesis of 2'-(4-methylumbelliferyl)-alpha-D-N-acetylneuraminic acid and detection of skin fibroblast neuraminidase in normal humans and in sialidosis. Biochemistry. 1979 Jun 26;18(13):2783–2787. doi: 10.1021/bi00580a014. [DOI] [PubMed] [Google Scholar]
- Wenger D. A., Sattler M., Clark C., Wharton C. I-cell disease: activities of lysosomal enzymes toward natural and synthetic substrates. Life Sci. 1976 Aug 1;19(3):413–420. doi: 10.1016/0024-3205(76)90047-3. [DOI] [PubMed] [Google Scholar]
- Willner J. P., Grabowski G. A., Gordon R. E., Bender A. N., Desnick R. J. Chronic GM2 gangliosidosis masquerading as atypical Friedreich ataxia: clinical, morphologic, and biochemical studies of nine cases. Neurology. 1981 Jul;31(7):787–798. doi: 10.1212/wnl.31.7.787. [DOI] [PubMed] [Google Scholar]