

Abstract
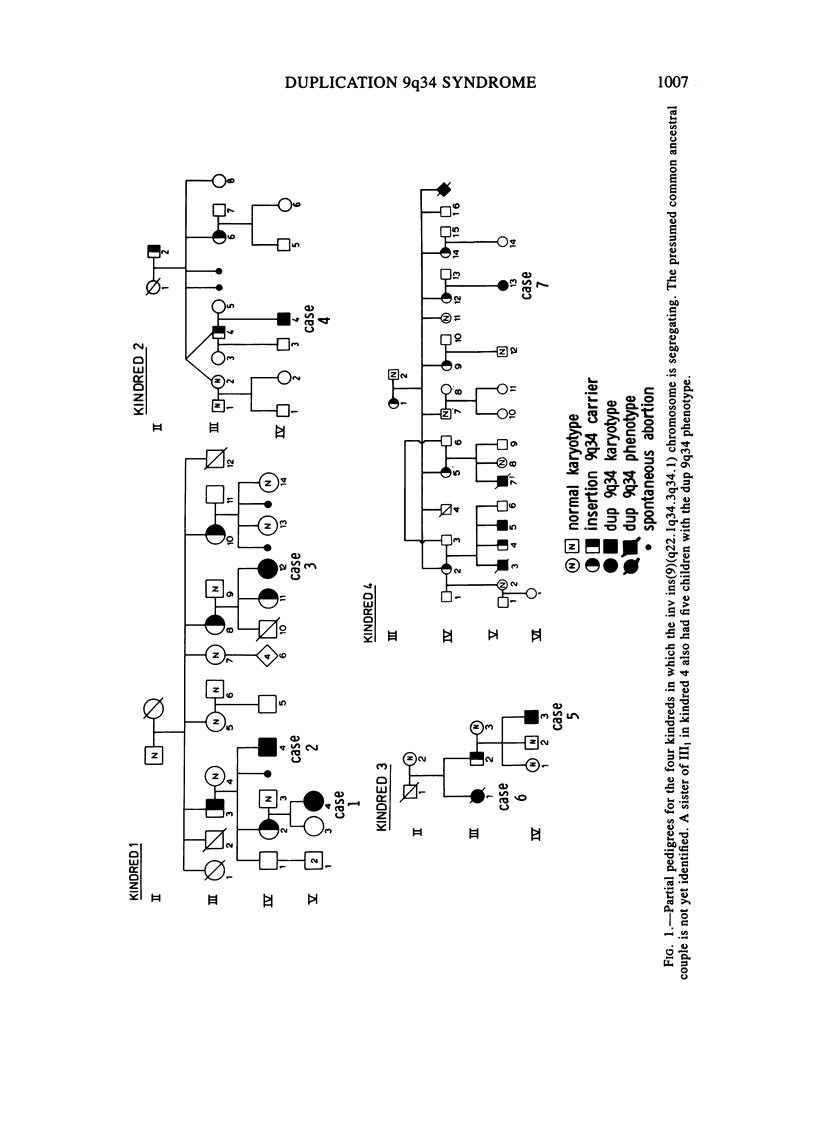
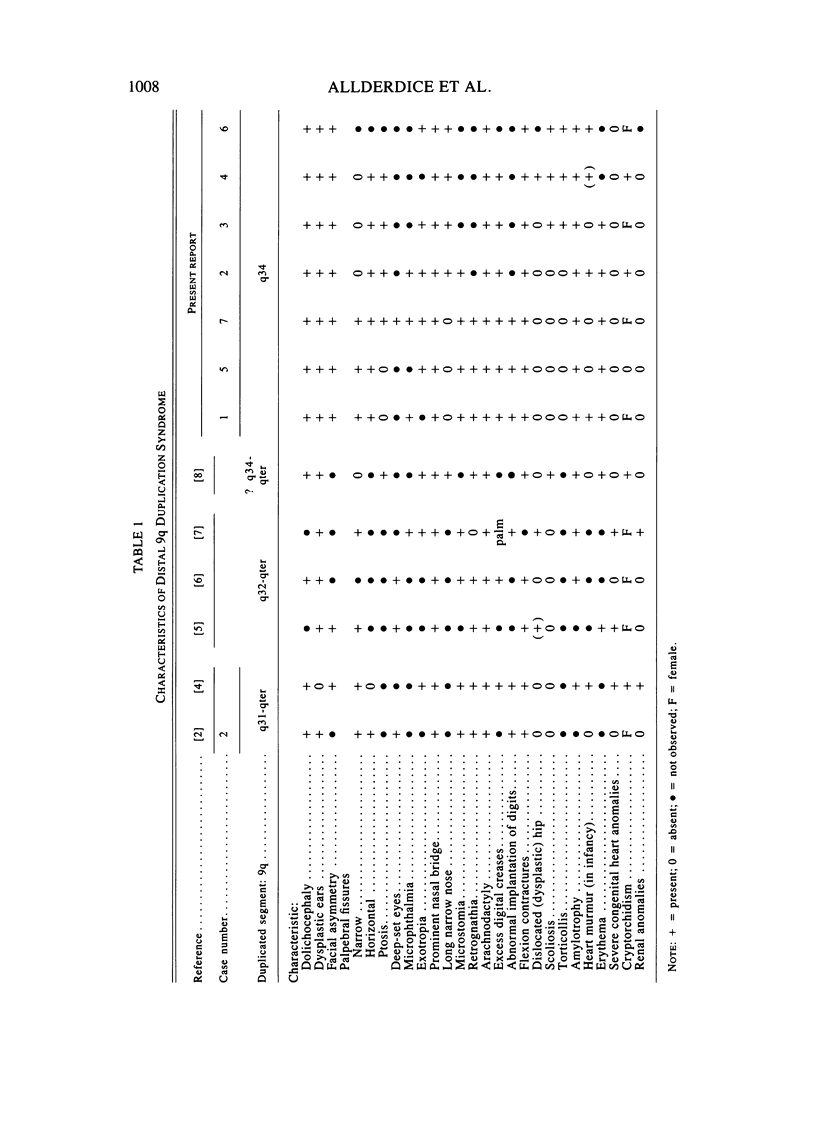
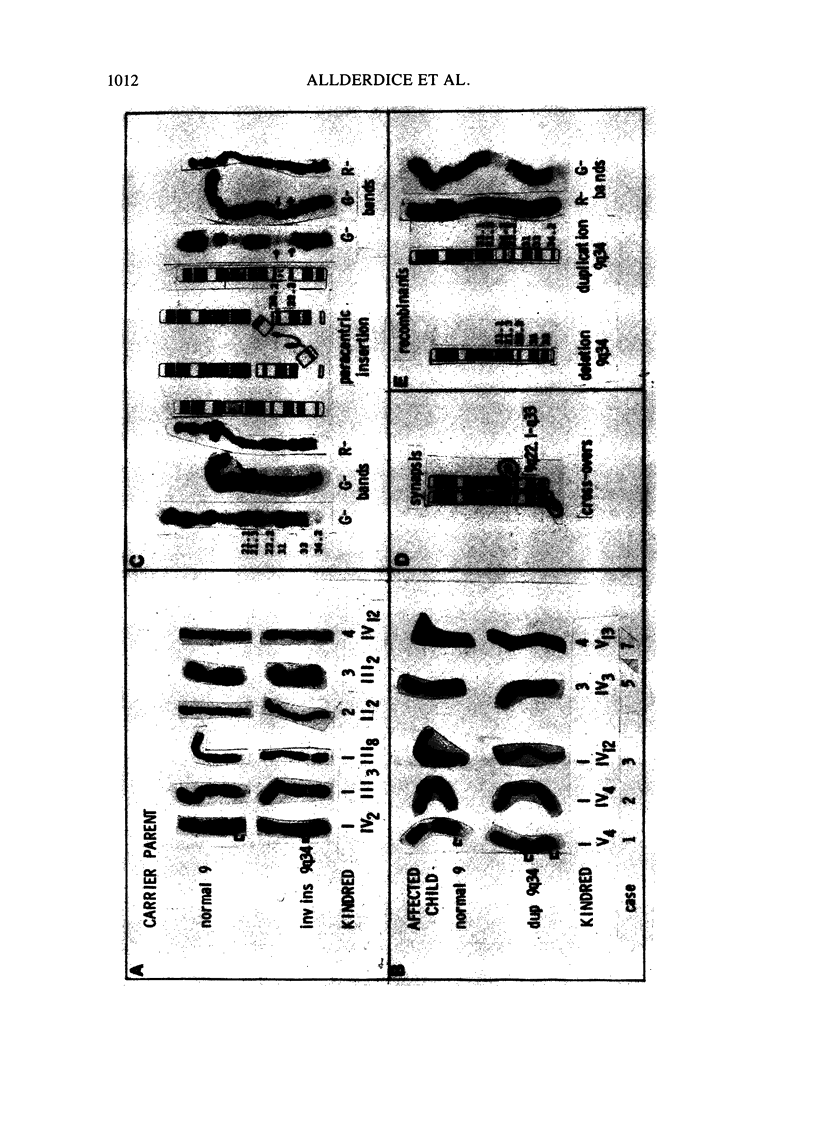
Phenotypic, karyotypic, and developmental homology between affected children of carriers of an inverted insertion (9) (q22.1q34.3q34.1) led to recognition of a new chromosome syndrome: dup 9q34. Individuals with dup 9q34 have slight psychomotor retardation, understand simple directions, and acquire a limited vocabulary. In childhood, many are hyperactive. Clinical features include low birth weight, normal birth length, and initial poor feeding and thriving. Musculo-skeletal systems are affected: there are joint contractures, long thin limbs, and striking arachnodactyly. There is abnormal implantation of the thumb, increased space between the first and second fingers, and excess digital creases. Marfan syndrome was a provisional diagnosis for several cases prior to cytogenetic analysis. Cardiovascular and ocular systems are minimally affected, erythema and heart murmurs occur, and ptosis and strabismus are frequent, but lens dislocation is not observed. Features at birth include: dolichocephaly, facial asymmetry, narrow horizontal palpebral fissures, microphthalmia, prominent nasal bridge, small mouth, thin upper lip with down-turned corners, and slight retrognathia. In older children, retrognathia is diminished and the nose becomes long and narrow. The new culture and chromosome banding techniques enable sorting of cases with the distal dup 9q phenotype into two groups. The cases with a longer dup 9q are more likely to develop with life-threatening congenital anomalies. The cases with the shorter dup 9q34 have a less severe long-term prognosis and will benefit, together with their parents, from special education. Female carriers of the inv ins(9) (q22.1q34.3q34.1) have about a 31% risk in each pregnancy to conceive a fetus affected by the dup 9q34 syndrome. A comparable figure is not yet available for male carriers.
Full text
PDF












Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Aftimos S. F., Hoo J. J., Parslow M. I. Partial trisomy 9q due to maternal 9/17 translocation. Am J Dis Child. 1980 Sep;134(9):848–850. doi: 10.1001/archpedi.1980.02130210032009. [DOI] [PubMed] [Google Scholar]
- Allderdice P. W., Browne N., Murphy D. P. Chromosome 3 duplication q21 leads to qter deletion p25 leads to pter syndrome in children of carriers of a pericentric inversion inv(3) (p25q21). Am J Hum Genet. 1975 Nov;27(6):699–718. [PMC free article] [PubMed] [Google Scholar]
- Mattei J. F., Mattei M. G., Ardissone J. P., Taramasco H., Giraud F. Pericentric inversion, inv(9) (p22 q32), in the father of a child with a duplication-deletion of chromosome 9 and gene dosage effect for adenylate kinase-1. Clin Genet. 1980 Feb;17(2):129–136. doi: 10.1111/j.1399-0004.1980.tb00121.x. [DOI] [PubMed] [Google Scholar]
- Pescia G., Jotterand-Bellomo M., de Crousaz H., Payot M., Martin D. Phénotype de la trisomie 9q distale chez un enfant présentant un chromosome surnuméraire remanié [t(X;9)]. Ann Genet. 1979;22(3):158–162. [PubMed] [Google Scholar]
- Pyeritz R. E., McKusick V. A. The Marfan syndrome: diagnosis and management. N Engl J Med. 1979 Apr 5;300(14):772–777. doi: 10.1056/NEJM197904053001406. [DOI] [PubMed] [Google Scholar]
- Subrt I., Janovský M., Jodl J. Partial trisomy 9q--chromosomal syndrome. Hum Genet. 1976 Oct 28;34(2):151–154. doi: 10.1007/BF00278883. [DOI] [PubMed] [Google Scholar]
- Turleau C., de Grouchy J., Chavin-Colin F., Roubin M., Brissaud P. E., Repessé G., Safar A., Borniche P. Partial trisomy 9q: a new syndrome. Humangenetik. 1975 Sep 23;29(3):233–241. doi: 10.1007/BF00297629. [DOI] [PubMed] [Google Scholar]