

Abstract
Targeted disruption of the pp60src (Src) gene has implicated this tyrosine kinase in osteoclast-mediated bone resorption and as a therapeutic target for the treatment of osteoporosis and other bone-related diseases. Herein we describe the discovery of a nonpeptide inhibitor (AP22408) of Src that demonstrates in vivo antiresorptive activity. Based on a cocrystal structure of the noncatalytic Src homology 2 (SH2) domain of Src complexed with citrate [in the phosphotyrosine (pTyr) binding pocket], we designed 3′,4′-diphosphonophenylalanine (Dpp) as a pTyr mimic. In addition to its design to bind Src SH2, the Dpp moiety exhibits bone-targeting properties that confer osteoclast selectivity, hence minimizing possible undesired effects on other cells that have Src-dependent activities. The chemical structure AP22408 also illustrates a bicyclic template to replace the post-pTyr sequence of cognate Src SH2 phosphopeptides such as Ac-pTyr-Glu-Glu-Ile (1). An x-ray structure of AP22408 complexed with Lck (S164C) SH2 confirmed molecular interactions of both the Dpp and bicyclic template of AP22408 as predicted from molecular modeling. Relative to the cognate phosphopeptide, AP22408 exhibits significantly increased Src SH2 binding affinity (IC50 = 0.30 μM for AP22408 and 5.5 μM for 1). Furthermore, AP22408 inhibits rabbit osteoclast-mediated resorption of dentine in a cellular assay, exhibits bone-targeting properties based on a hydroxyapatite adsorption assay, and demonstrates in vivo antiresorptive activity in a parathyroid hormone-induced rat model.
Protein tyrosine kinases (1) and phosphatases (2) have been implicated in mediating a host of intracellular activities, including cell proliferation, migration, and differentiation. Many such proteins are able to selectively bind their cognate targets and initiate a cascade of signaling events through key modular domains that control protein–protein interactions (3). One such domain, the Src homology 2 (SH2) domain, has been determined to play a pivotal role in many signaling pathways by recognizing phosphotyrosine (pTyr) sequences of cognate proteins (4). The SH2 domain of the nonreceptor protein tyrosine kinase Src has been shown to interact with focal adhesion kinase, p130cas, p85, phosphatidylinositol 3-kinase, and p68sam (5–8). Small molecules designed to inhibit SH2-mediated protein–protein interactions have promise as pharmaceutical agents to block specific intracellular pathways critically involved in the pathogenesis of certain diseases. Targeted disruption of the Src gene in mice has revealed a critical role of Src in the normal function of osteoclasts (9). Osteoclasts isolated from src−/− mice show abnormal cellular morphology and impaired functional properties, including an inability to form an organized actin cytoskeleton at the site of adhesion to bone matrix, as well as the so-called ruffled border, which ultimately result in their lack of bone-resorbing activity (10, 11). Interestingly, despite the ubiquitous nature of Src in many cell types, src−/− mice display osteopetrosis as the only observable phenotype (9). This finding suggests the existence of overlapping, compensatory signaling pathways in unaffected cell types, presumably by other Src family members, and furthermore, that a small-molecule inhibitor of Src function would be particularly well suited as an antiresorptive agent.
To date, most Src SH2 inhibitors have been phosphopeptides (12) or peptidomimetics (13, 14), which incorporate pTyr or pTyr-like mimics. pTyr-containing inhibitors suffer from poor transport properties and are rapidly degraded by phosphatases. Unfortunately, attempts to design metabolically stable pTyr mimics generally have resulted in compounds with weaker affinity for the target SH2 domain (15). Consequently, one of our objectives was to design metabolically stable pTyr mimics with Src SH2 binding affinity comparable to pTyr. Additionally, we required that the designed pTyr mimic target bone (hydroxyapatite), thereby conferring tissue selectivity for osteoclasts and reducing undesired effects in other cell types. Finally, we sought to replace the post-pTyr sequence of Ac-pTyr-Glu-Glu-Ile (1) with a nonpeptide template demonstrating increased affinity for Src SH2. We report here the structure-based design of a series of compounds that fulfill these criteria. We have synthesized a large set of Src SH2 inhibitors to systematically explore chemical diversity within this series of molecules. The work here highlights the lead compound, AP22408, in terms of its Src binding, bone targeting, and antiresorptive properties in an osteoclast-mediated cellular assay and an in vivo thyroparathyroidectomized (TPTX) model of parathyroid hormone-induced bone resorption.
Materials and Methods
Chemical Synthesis.
The synthesis of AP22408 in terms of its bicyclic template and pTyr mimic, 3′,4′-diphosphonophenylalanine (Dpp), is shown in Figs. 4 and 5, respectively. A negative control compound, AP22409, was prepared from a Dpp intermediate (5) by coupling with benzylamine. AP22650 was synthesized from known 4′-phosphonophenylalanine (16) by coupling with amine 4 and further modified according to Fig. 4. Full experimental details will be described elsewhere.
Figure 4.
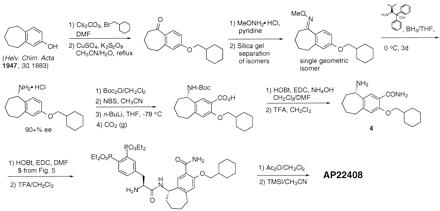
Chemical synthesis of amine intermediate 4 and AP22408. DMF, dimethylformamide; BH3/THF, borane-tetrahydrofuran; Boc2O di-tert-butyl dicarbonate; Boc, tert-butoxycarbonyl; NBS, N-bromosuccinimide; n-BuLi, n-butyllithium; THF, tetrahydrofuran; CO2, carbon dioxide; HOBt, 1-hydroxybenzotriazole; EDC, 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride; TFA, trifluoroacetic acid; Ac2O, acetic anhydride; TMSI, iodotrimethylsilane; Me, methyl; Et, ethyl.
Figure 5.
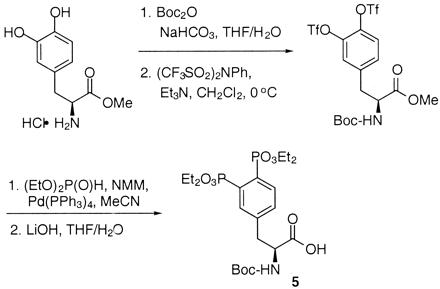
Chemical synthesis of Dpp intermediate 5. Tf, trifluoromethanesulfonyl; Boc, tert-butoxycarbonyl; NMM, 4-methylmorpholine; Me, methyl; Et, ethyl; Ph, phenyl.
Molecular Modeling.
When this work was initiated, no high-resolution crystal structures of Src SH2 were available for docking ligands that span the entire binding site. Therefore, we used a high-resolution (1.0 Å) crystal structure of Lck SH2 complexed with the phosphopeptide 1 (Fig. 3) (17). We have successfully exploited this model in the design of highly potent Src SH2 inhibitors and in the accurate prediction of their binding mode before structural determination (x-ray and NMR). Where relevant, to compare Lck with Src, residue numbers for Src are given in parenthesis. The binding site model includes all residues within 7 Å of the ligand. During conformational searching and energy minimization, all residues were held rigid except those known to exhibit conformational changes upon binding to different ligands. Residues Lys-182, Ile-193, Ser-194, Arg-193, and Leu-216 are allowed to move freely during optimization whereas residues Glu-155, Ser-156, Glu-157, Ser-158, Glu-159, and Ser-164 were subjected to a flat well potential of radius 0.5 Å and a constraint of 20.0 kJ/Å2 outside this radius. Docking was carried out by using the mcdock conformational searching/energy minimization procedure of flo96 (18, 19). Each compound was subjected to 5,000 cycles of conformational searching and energy minimizations to identify conformations most likely to bind to the SH2 domain. Each cycle involved 400 rapid Monte Carlo search steps followed by energy minimization of the best conformer from the set of 400. The program returned the 25 lowest energy conformations found for visual inspection. Docked molecules were evaluated by using the following criteria: steric fit, hydrophobic and hydrogen bonding interactions, low molecular mechanics energy, and low internal ligand energy.
Figure 3.
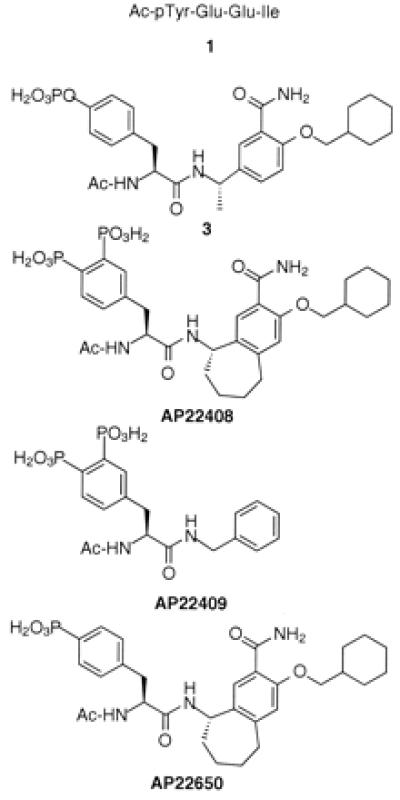
Chemical structures of 1, 3, AP22408, AP22409, and AP22650.
X-Ray Crystallography.
Crystals of Lck (S164C) SH2 complexed with compound AP22408 were obtained from PEG 4000 with Tris buffer. Diffraction data were collected at −160°C with a Rigaku area detector. The crystals are orthorhombic, space group P212121 with unit cell a = 44.87, b = 56.26, and c = 102.77 Å with two molecules in the asymmetric unit. Diffraction data extended to 2.4-Å resolution. The structure was determined by molecular replacement using high-resolution Lck SH2-phosphopeptide (pTyr-Glu-Glu-Ile) as a model. Electron density for Lck (S164C) SH2 complex with AP22408 was as expected, however, the BC loop of the protein was not fully defined and required some rebuilding. The structure was refined by simulated annealing using x-plor. Parameters of the complex included Rfree = 0.36 and r = 0.23. The model contained 108 protein residues, AP22408, and 40 water molecules.
Hydroxyapatite Chromatography Assay.
Retention times were measured on a high-pressure hydroxyapatite adsorption chromatography column (TosoHaas TSK-Gel HA 1000 7.5 mm × 75 mm) using a Hewlett–Packard HP1050 series chromatograph with a variable wavelength UV detector. Compounds were loaded in 10 mM sodium phosphate, 150 mM sodium chloride, pH 6.8 and eluted with a linear gradient of 10–500 mM sodium phosphate, 150 mM sodium chloride, pH 6.8.
The retention time of the compound was expressed in terms of K = (tR − tV)/tV, where tR is the measured retention time and tV is the void volume of the column. This K value was corrected to give K′ values by using two reference compounds to compensate for intercolumn variations.
In Vitro Assays.
Details of both the Src SH2 binding assay (20, 21) and the osteoclast-mediated bone resorption assay (21) have been described.
In Vivo TPTX Rat Model of Bone Resorption.
Female Wistar rats (Charles River Laboratories, 201–225 g) underwent surgical removal of the thyroid and parathyroid glands on day 0 and were immediately started on a pair-fed, low-calcium diet (Harlan Teklad TD 96965, ≤0.003% calcium, ≤0.04% phosphate). The success of the surgical TPTX was confirmed on day 2 by obtaining blood and checking for a decrease in the serum calcium level. Animals were eligible for entry into the study if the serum calcium level was < 7 mg/dl. Eligible animals were randomly assigned to receive AP22408 or vehicle control (n = 6/group) beginning on day 3. Before compound administration on day 3, a blood sample was obtained via the jugular vein for baseline serum calcium determination. AP22048 (50 mg/kg twice a day) or vehicle (4 ml/kg, 0.9% saline, twice a day) was administered i.v. by tail vein injection for 4 consecutive days. After the first dose on day 3, parathyroid hormone (bovine, 1–34, 9.4 μM vehicle)-charged ALZET (ALZA, model 2002, 0.5 μl/h, 14 d) osmotic minipumps were implanted s.c. Blood was collected for serum calcium in the morning of each subsequent day through day 18. On treatment days, blood was collected before the first i.v. treatment for the day. Serum calcium concentration was measured by a commercially available, colorimetric assay (Sigma, no. 588–3) that was modified for use in a microtiter format.
The null hypothesis that treatment fails to alter repeated serum calcium measures was assessed by using multivariate ANOVA (sas version 6.12). Data from days 5–18 were used in this analysis and were “smoothed” by using a moving average with a window of three units. The areas under the treatment-specific serum calcium vs. time curves were estimated by using a mixed model, which accounts for within-animal correlations and random effects, and compared. The two-tailed significant level was set at P < 0.05.
Results
Structure-Based Design of AP22408.
X-ray structures of the Src and Lck SH2 domains complexed with the cognate phosphopeptide, Ac-pTyr-Glu-Glu-Ile (1, Fig. 3) have yielded detailed information of key molecular interactions between the pTyr and the pTyr-binding pocket of the proteins (22, 23). Recently, we made the unexpected observation that the crystal structure of Src-SH2, when crystallized from citrate buffer, contained a well-defined citrate ion in the pTyr binding pocket (M.H., unpublished results). Similar to the phosphate group of pTyr from known peptide complexes with either Src or Lck SH2 domains, the citrate forms numerous ionic and hydrogen bonds with both protein backbone and side-chain atoms. These include ionic interactions with the conserved Arg-158 and Arg-178 residues, as well as hydrogen bonds with Ser-180, Thr-182 and the backbone NH of Glu-181 (Fig. 1A). Importantly, the citrate complex with Src SH2 revealed molecular interactions not previously observed in SH2-phosphopeptide cocrystal structures. Specifically, this included an ionic bond between citrate and Lys-206 and an intermolecular hydrogen bond with the backbone NH of Thr-182. One of our goals was to leverage these additional molecular interactions in the design of novel pTyr mimics that would be both hydrolytically stable and osteoclast-selective by virtue of bone-targeting properties.
Figure 1.
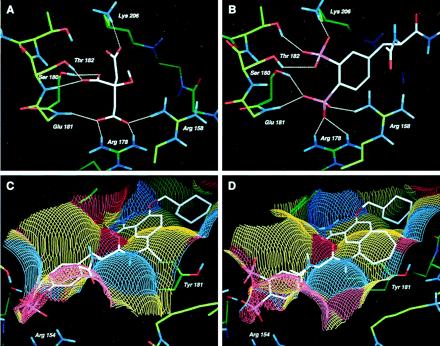
Structure-based design of AP22408. (A) X-ray crystal structure of citrate complexed to the pTyr-binding pocket of Src SH2 depicting hydrogen bonding atoms. (B) Model of Dpp in the pTyr binding pocket of Src SH2, highlighting the hydrogen bonding network. (C) Model of 3 complexed with Lck SH2, illustrating the solvent accessible surface detailed in terms of both hydrophobic and hydrogen bonding interactions. Yellow colors indicate hydrophobic surfaces, blue colors indicate hydrogen bond accepting surfaces, and red colors indicate hydrogen bond donating surfaces. (D) Model of AP22408 in Lck SH2 highlighting the hydrophobic interactions of the bicyclic template.
A detailed analysis of the Src SH2-citrate cocrystal structure led to the design of several pTyr mimics, including Dpp (see Fig. 2). To investigate the mode of binding, Dpp was flexibly docked into the pTyr-binding pocket (pY site) of the Src SH2 model. The results indicated that the 3′-phosphonate group of Dpp would form both ionic and hydrogen bonds with Lys-206 and Thr-182 (Fig. 1B). The 4′-phosphonate group of Dpp was predicted to interact with Arg-158, Arg-178, and Ser-180. When compared with pTyr from the Lck SH2-phosphopeptide cocrystal structures, the 4′-phosphonate moiety of Dpp does not penetrate as deeply into the pY site and was predicted to form a different set of molecular interactions. Nonetheless, the Dpp moiety was expected to be functionally equivalent to pTyr and, more importantly, to provide a metabolically stable (e.g., phosphatase-resistant) pTyr mimic, as well as confer osteoclast selectivity by virtue of its bone-targeting (hydroxyapatite binding) properties.
Figure 2.
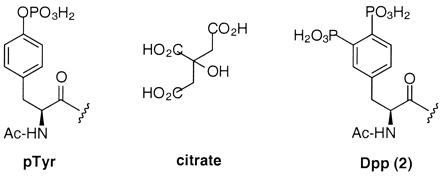
Chemical structures of pTyr, citrate, and Dpp (2).
To further achieve a pharmacologically effective small molecule, we simultaneously pursued a nonpeptidic template to replace the Glu-Glu-Ile sequence of phosphopeptide 1. In this regard, a reported nonpeptide template developed for the Src SH2 domain (13, 14) provided a prototype for the structure-based design of our bicyclic template (see below). Relative to the nonpeptide 3 (Fig. 3), a model of it complexed with Lck SH2 (Fig. 1C) revealed several important molecular interactions: (i) the central phenyl ring of 3 stacks perpendicular to the phenyl ring of Tyr-181; (ii) the primary benzamide group of 3 forms two hydrogen bonds with the protein; and (iii) the cyclohexane ring extends deeply into the Ile-binding pocket (pY+3 site), forming numerous hydrophobic contacts. An analysis of this model revealed that the central aromatic ring of 3 did not extend significantly beyond the phenyl ring of Tyr-181. Therefore, a molecule that would more effectively complement the hydrophobic surface of the Src SH2 (see Fig. 1C) might afford higher binding affinity. Modeling suggested that a seven-membered ring fused directly to the central ring of 3 would accommodate the complete Src SH2 hydrophobic architecture. The resultant bicyclic benzamide was advanced as a promising nonpeptide template for use in concert with the Dpp moiety (Fig. 1D) to create AP22408 (Fig. 3) and analogs thereof.
After systematically exploring the structure activity relationships of this series, AP22408 was rapidly identified as a lead compound. The work here focuses specifically on AP22408, the negative control compound AP22409, and a nonbone-targeted analog AP22650 (Fig. 3). AP22409 also contains a Dpp moiety (and hence should target bone) but was expected to bind weakly to Src SH2 because of the deletion of the three constituent groups (i.e., fused cycloaliphatic ring, amide, and a pY+3 cyclohexylmethyloxy substituent) relative to the bicyclic template of AP22408. For AP22650, the replacement of Dpp by 4′-phosphonophenylalanine was expected to provide high binding affinity to Src SH2 but result in a complete loss in bone-targeting property (hence, a negative control with respect to cellular studies on osteoclasts). The synthesis of AP22408 is generally outlined in Figs. 4 and 5; a detailed chemistry discussion, as well as other structure-activity studies, will be reported elsewhere.
Src SH2 Binding of AP22408.
A fluorescence polarization-based competitive binding assay was used to determine the IC50 values for compound binding to the Src SH2 domain (20, 21). Compounds 1 and 3 were found to bind Src SH2 with IC50 values of 5.5 μM and 2.2 μM, respectively (Table 1). AP22408 inhibited Src SH2-ligand binding with an IC50 of 0.30 μM. This represented significantly increased binding affinity relative to both 1 and 3 and shows AP22408 to be the tightest binding inhibitor for Src SH2 reported to date. AP22650 binds to Src SH2 with an IC50 of 1.3 μM, which suggests that the approximately 4-fold higher affinity of AP22408, relative to AP22650, may be caused by the additional molecular interactions of the 3′-phosphonate moiety of Dpp within the pTyr binding pocket. As predicted, AP22409 did not show Src SH2 binding (IC50 > 500 μM).
Table 1.
Comparative Src SH2 binding affinities (IC50s) for compounds 1, 3, AP22408, AP22409, and AP22650 using a competitive fluorescence polarization assay
| Compound | Src (IC50 μM) |
|---|---|
| 1 | 5.56 ± 0.21 |
| 3 | 2.2 ± 0.09 |
| AP22408 | 0.30 ± 0.06 |
| AP22650 | 1.3 ± 0.16 |
| AP22409 | >500 |
X-Ray Structure of AP22408 Complexed with Lck (S164C) SH2 Domain.
To confirm our predictions for the binding mode of AP22408, we determined its structure as a cocrystal with Lck (S164C) SH2 (Fig. 6) to a resolution of 2.4 Å. The experimentally determined bound conformation proved to be in good agreement with that predicted by molecular modeling (see Fig. 1 B–D for detailed molecular interactions and Fig. 6 for an overlay of the model and x-ray structures). The x-ray structure confirmed that the Dpp group of AP22408 binds in the pTyr pocket. The oxygens of the 4′-phosphonate of Dpp form ionic interactions with Arg-154 (for Src, Arg-178), and hydrogen bonds with the backbone NH of Glu-157 (Glu-181) and backbone NH of Ser-158 (Thr-182). The 3′-phosphonate of Dpp forms ionic interactions with Lys-182 (Lys-206) and a hydrogen bond with Ser-158 (Thr-182). As predicted, the seven-membered ring of the bicyclic template fits well in the hydrophobic groove formed by the side chains of Lys-179 (Lys-206) and Tyr-181 (Tyr-205). The benzamide carbonyl forms a hydrogen bond with the backbone NH of Lys-182 (Lys-206), displacing one of the two water molecules observed in the phosphopeptide complex. The second water molecule is not displaced and is hydrogen-bonded to the benzamide NH group of AP22408 and the backbone carbonyl of Ile-193 (Ile-217). Finally, as predicted, the cyclohexyl group of AP22408 extends into the hydrophobic pY+3 pocket.
Figure 6.
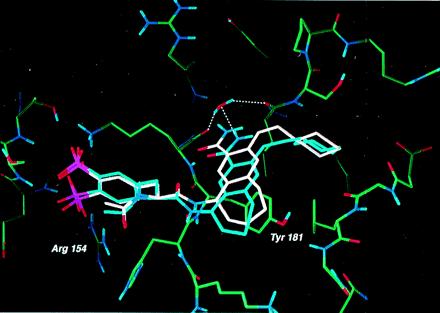
Superposition of the model of AP22408 (white) and the x-ray (blue) crystal structure of the AP22408-Lck SH2 (S164C) complex. The water-mediated hydrogen bonds (determined crystallographically, blue) between the carboxamide of AP22408 and the protein were not predicted as no water molecules were included in our model.
Bone-Targeting Properties of AP22408 as Determined from Hydroxyapatite Chromatography.
To evaluate the ability of bone-targeted compounds such as AP22408 to bind to bone, a hydroxyapatite adsorption chromatography assay was developed (see Materials and Methods). The hydroxyapatite chromatography results show that pTyr-containing compounds have essentially no affinity for hydroxyapatite (K′< 0.1). Likewise, AP22650, which contains a single 4′-phosphonate as its pTyr mimic, does not bind significantly to hydroxyapatite (K′< 0.1). As reference compounds, alendronate (a bone-targeted bisphosphonate drug) gave a K′ value of 3.6, and tetracycline (a compound known to associate with bone) gave a K′ value of 2.0. The K′ value determined for AP22408 was 1.9, thus indicating that it exhibits bone-targeting properties. This property was directly related to the Dpp moiety of AP22408 as supported by additional studies in which functionalities on the molecule were systematically deleted (data not shown). We also have shown that [3H]AP22408 and [3H]AP22409, when incubated with dentine, bind in a concentration- and time-dependent manner to further support the bone-targeting design of this novel pTyr mimic (S.V., unpublished results).
Osteoclast-Mediated Antiresorptive Activity of AP22408.
AP22408, AP22409, and AP22650 were assayed for their ability to inhibit rabbit osteoclast-mediated resorption of dentine slices. These compounds were dosed in two protocols that differed only with regard to whether the dentine was preincubated with compound or not before addition of the osteoclasts to the dentine (Table 2). Preincubation should allow the bone-targeted compounds to selectively target and accumulate on the dentine surface, thus exposing the resorbing osteoclasts to a higher concentration of substrate. AP22408 and AP22409 demonstrated IC50 values with preincubation of 1.6 μM and >200 μM, respectively. When dosed subsequent to adhesion of the osteoclasts, AP22408 had a significantly higher IC50 value of 57.0 μM (AP22409 > 200 μM). Neither of these compounds demonstrated toxicity at any of the concentrations tested as monitored by the presence of tartrate-resistant acid phosphatase-positive cells or surrounding fibroblasts. Conversely, AP22650, a molecule with similar affinity for Src SH2 but devoid of bone-targeting properties, was inactive in the osteoclast assay (IC50 >200 μM). Taken together, these data strongly suggest that AP22408 targets bone through the Dpp moiety and that such a property translates into cellular efficacy with respect to the bone-resorbing osteoclast model described above.
Table 2.
Comparative inhibition properties of AP22408, AP22409, and AP22650 in a rabbit osteoclast-mediated resorption of dentine assay
| Compound | IC50 (μM) (+/− preincubation)
|
|
|---|---|---|
| + | − | |
| AP22408 | 1.6 | 57.0 |
| AP22409 | >200 | >200 |
| AP22650 | >200 | >200 |
In Vivo TPTX Rat Antiresorptive Activity of AP22408.
The TPTX model is an established animal model for in vivo evaluation of antiresorptive compounds (24, 25). The time course of effect on serum calcium concentration is shown in Fig. 7 for animals receiving the vehicle control versus AP22408. The effect of AP22408 was evident beginning at day 5, the second day of treatment with AP22408. There was a clear separation of control and AP22408-treated groups from day 5 through the end of the observation (day 18). The area under the curve analysis indicates statistically significant (P = 0.0379) differences between the two treatments. Animals treated with the negative control (AP22409) demonstrated serum calcium levels comparable to the vehicle control animals (data not shown). These data demonstrate that AP22408 provides a significant beneficial antiresorptive effect in this well-characterized animal model of parathyroid hormone-induced bone resorption.
Figure 7.
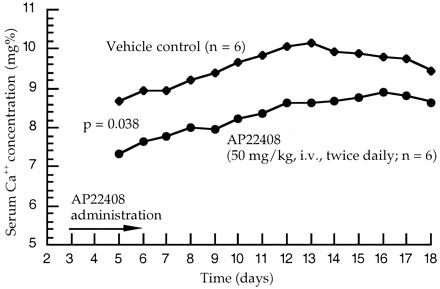
In vivo antiresorptive activity of AP22408 in TPTX rats. Mixed model area under the curve analysis: three point moving average data (days 5–18).
Discussion
Src is composed of five structural modules: a unique region, SH3 and SH2 domains, a catalytic domain, and a C-terminal tail, which is capable of intramolecular interaction with the SH2 domain when phosphorylated at Tyr-527. Although the catalytic domain of Src is important for both autophosphorylation and downstream phosphorylation of substrate proteins, the importance of the SH2 domain in regulating Src-dependent intracellular signaling processes has not been fully elucidated. It has been postulated that in particular signaling processes, such as integrin-mediated signaling, the SH2 domain acts as an adaptor protein, recruiting specific proteins to the signaling complex (26, 27). This hypothesis is supported by studies that have shown that coexpression of mutated or truncated Src (modifications anticipated to render catalytically inactive protein) in src −/− osteoclasts appear to partially rescue the osteopetrosis phenotype and cellular activity (11, 28).
The challenge of designing small-molecule inhibitors specific for Src SH2 derive from the highly homologous nature of this domain within the Src family (29, 30). The observation that Src is the only member in the family to contain a cysteine residue proximal to the 3′ position of pTyr has led to an initial series of Src inhibitors designed to target this residue (21, 31–33). These inhibitors capture this residue by incorporating an aldehyde in the 3′ position of pTyr (or substituted phenylalanine derivatives). In limited cases, inhibition of osteoclast activity has been observed and provides impetus to drug discovery focused on Src for osteoporosis (21).¶
As an approach to the discovery of Src SH2 inhibitors, we describe the structure-based design of nonhydrolyzable pTyr mimics, which simultaneously provide molecular recognition (Src SH2 binding) and bone-targeting (osteoclast selectivity). In fact, we have developed a series of pTyr mimics having such dual functional properties. Here, we disclose the Dpp moiety as both a pTyr replacement and bone-targeting chemical group. The concurrent structure-based optimization of a nonpeptide template to replace the cognate phosphopeptide sequence to bind the Src SH2 domain (i.e., pY+1, pY+2, and pY+3 sites) by a novel bicyclic benzamide template provided the opportunity to advance an additional class of Src inhibitors. Specifically, we show AP22408 is one of the tightest binding, small-molecule Src SH2 inhibitors reported to date. An x-ray structure of AP22408 cocrystallized with Lck (S164C) SH2 confirms the design concepts underlying this additional series of Src inhibitors.
AP22408, by virtue of the Dpp moiety, demonstrates a remarkably strong affinity for bone and has been shown to both accumulate at elevated levels on the bone surface and effect potent inhibition of osteoclast-mediated bone resorption. A direct comparison of AP22408 with two negative control compounds, AP22409 (a bone-targeted analog that does not bind to Src SH2) and AP22650 (a nonbone-targeted analog that has high affinity to bind Src SH2), in an rabbit osteoclast resorption assay validate the mechanism of action and biological properties of AP22408. We also have examined AP22408 in a series of mechanism-based cellular assays to correlate its in vitro, antiresorptive activity with binding to Src SH2 (S.V., unpublished results). Here, we further demonstrate that AP22408 is an inhibitor of Src SH2 that shows a statistically significant antiresorptive activity in an in vivo TPTX model of parathyroid hormone-induced bone resorption. Collectively, these data strongly support the concept that Src is intimately involved in signaling pathways involved with bone resorption by osteoclasts and further validates Src as a promising therapeutic target for the treatment of osteoporosis as well as other bone diseases such as Paget's disease, osteolytic bone metastasis, and hypercalcemia associated with malignancy.
Acknowledgments
We thank the protein biochemistry, assay development, and cell biology groups of ARIAD Pharmaceuticals for their various considerable contributions. W.S. thanks Rick Brawley for graphics assistance.
Abbreviations
- SH2
Src homology 2
- pTyr
phosphotyrosine
- Dpp
3′,4′-diphosphonophenylalanine
- TPTX
thyroparathyroidectomized
Footnotes
Data deposition: Atomic coordinates have been deposited in the Protein Data Bank (PDB ID code 1FBZ).
Dunnington, D., Votta, B., Hand, A., Appelbaum E., Jones, C., Prichett, W., Holt, D., Yamashita, D. & Gowen, M. (1996) Annual Meeting of the American Society of Bone and Mineral Research, Sept. 8–11, 1996, Seattle, WA, poster no. 395.
References
- 1.Levitzki A. Pharmacol Ther. 1999;82:231–239. doi: 10.1016/s0163-7258(98)00066-7. [DOI] [PubMed] [Google Scholar]
- 2.Tonks N K. Adv Pharmacol. 1996;36:91–119. doi: 10.1016/s1054-3589(08)60578-5. [DOI] [PubMed] [Google Scholar]
- 3.Cohen G B, Ren R, Baltimore D. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 4.Thomas S M, Brugge J S. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 5.Fukui Y, Hanafusa H. Mol Cell Biol. 1991;11:871–874. doi: 10.1128/mcb.11.4.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaller M D, Hilderbrand J D, Shannon J D, Fox J W, Vines R R, Parsons J T. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor S J, Shalloway D. Nature (London) 1994;368:867–871. doi: 10.1038/368867a0. [DOI] [PubMed] [Google Scholar]
- 8.Petch L A, Bockholt S M, Bouton A, Parsons J T, Burridge K. J Cell Sci. 1995;108:1371–1379. doi: 10.1242/jcs.108.4.1371. [DOI] [PubMed] [Google Scholar]
- 9.Soriano P, Montgomery C, Geske R, Bradley A. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 10.Boyce B F, Yoneda T, Lowe C, Soriano P, Mundy G R. J Clin Invest. 1992;90:1622–1627. doi: 10.1172/JCI116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzberg P L, Xing L, Hoffmann O, Lowell C A, Garrett L, Boyce B F, Varmus H E. Genes Dev. 1997;11:2835–2844. doi: 10.1101/gad.11.21.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawyer T K. Biopolymers. 1998;47:243–261. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 13.Lunney E A, Para K S, Rubin J R, Humblet C, Fergus J H, Marks J S, Sawyer T K. J Am Chem Soc. 1997;119:12471–12476. [Google Scholar]
- 14.Lunney, E. A., Para, K. S., Plummer, M. S., Prasad, J. V. N. V., Saltiel, A. R., Sawyer, T. & Shahripour, A. (1997) PCT Intl. Patent W097/12903.
- 15.Botfield M C, Green J. Annu Rep Med Chem. 1995;30:227–237. [Google Scholar]
- 16.Thurieau C, Simonet S, Paladino J, Prost J-F, Verbeuren T, Fauchére J-L. J Med Chem. 1994;37:625–629. doi: 10.1021/jm00031a012. [DOI] [PubMed] [Google Scholar]
- 17.Tong L, Warren T C, King J, Betageri R, Rose J, Jakes S. J Mol Biol. 1996;256:601–610. doi: 10.1006/jmbi.1996.0112. [DOI] [PubMed] [Google Scholar]
- 18.McMartin C, Bohacek R S. J Comput Aided Mol Design. 1997;11:333–344. doi: 10.1023/a:1007907728892. [DOI] [PubMed] [Google Scholar]
- 19.ThistleSoft, Inc. flo96. Colebrook, CT: ThistleSoft; 1996. [Google Scholar]
- 20.Lynch B A, Loiacono K A, Tiong C L, Adams S E, Macneil I A. Anal Biochem. 1997;247:77–82. doi: 10.1006/abio.1997.2042. [DOI] [PubMed] [Google Scholar]
- 21.Violette S M, Shakespeare W C, Bartlett C, Guan W, Smith J A, Rickles R J, Bohacek R S, Holt D A, Baron R, Sawyer T K. Chem Biol. 2000;7:225–235. doi: 10.1016/s1074-5521(00)00090-9. [DOI] [PubMed] [Google Scholar]
- 22.Eck M J, Shoelson S E, Harrison S C. Nature (London) 1993;362:87–91. doi: 10.1038/362087a0. [DOI] [PubMed] [Google Scholar]
- 23.Waksman G, Shoelson S E, Pant N, Cowburn D, Kuriyan J. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 24.Frost H M, Lee W S S. Bone Miner. 1992;18:227–236. doi: 10.1016/0169-6009(92)90809-r. [DOI] [PubMed] [Google Scholar]
- 25.Green J R, Müller K, Jaeggi K A. J Bone Miner Res. 1994;9:745–751. doi: 10.1002/jbmr.5650090521. [DOI] [PubMed] [Google Scholar]
- 26.Schwartzberg P. Oncogene. 1998;17:1463–1468. doi: 10.1038/sj.onc.1202176. [DOI] [PubMed] [Google Scholar]
- 27.Klinghoffer R A, Sachsenmaier C, Cooper J A, Soriano P. EMBO J. 1999;18:2459–2471. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlaepfer D D, Broome M A, Hunter T. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Superti-Furga G, Courtneidge S A. BioEssays. 1995;17:321–330. doi: 10.1002/bies.950170408. [DOI] [PubMed] [Google Scholar]
- 30.Waksman G, Kominos D, Robertson S C, Pant N, Baltimore D, Birge R B, Cowburn D, Hanafusa H, Mayer B J, Overduin M, et al. Nature (London) 1992;358:646–653. doi: 10.1038/358646a0. [DOI] [PubMed] [Google Scholar]
- 31.Shakespeare W C, Bohacek R S, Narula S S, Azimioara M D, Yuan R W, Dalgarno D C, Madden L, Botfield M C, Holt D A. Bioorg Med Chem Lett. 1999;9:3109–3112. doi: 10.1016/s0960-894x(99)00545-4. [DOI] [PubMed] [Google Scholar]
- 32.Charifson P S, Shewchuk L M, Rocque W, Hummel C W, Jordan S R, Mohr C, Pacofsky G J, Peel M R, Rodriguez M, Sternbach D D, Counsler T G. Biochemistry. 1997;36:6283–6293. doi: 10.1021/bi970019n. [DOI] [PubMed] [Google Scholar]
- 33.Alligood K J, Charifson P S, Crosby R, Consler T G, Feldman P L, Gampe R T, Gilmer T M, Jordan S R, Milstead M W, Mohr C, Peel M R, et al. Bioorg Med Chem Lett. 1998;7:1189–1194. doi: 10.1016/s0960-894x(98)00195-4. [DOI] [PubMed] [Google Scholar]