

Abstract
This study was designed to elucidate the mechanism underlying the inhibition of endothelial cell growth by laminar shear stress. Tumor suppressor gene p53 was increased in bovine aortic endothelial cells subjected to 24 h of laminar shear stress at 3 dynes (1 dyne = 10 μN)/cm2 or higher, but not at 1.5 dynes/cm2. One of the mechanisms of the shear-induced increase in p53 is its stabilization after phosphorylation by c-Jun N-terminal kinase. To investigate the consequence of the shear-induced p53 response, we found that prolonged laminar shear stress caused increases of the growth arrest proteins GADD45 (growth arrest and DNA damage inducible protein 45) and p21cip1, as well as a decrease in phosphorylation of the retinoblastoma gene product. Our results suggest that prolonged laminar shear stress causes a sustained p53 activation, which induces the up-regulation of GADD45 and p21cip1. The resulting inhibition of cyclin-dependent kinase and hypophosphorylation of retinoblastoma protein lead to endothelial cell cycle arrest. This inhibition of endothelial cell proliferation by laminar shear stress may serve an important homeostatic function by preventing atherogenesis in the straight part of the arterial tree that is constantly subjected to high levels of laminar shearing.
Hemodynamic forces regulate the structure and function of the blood vessel wall (1, 2). Vascular endothelial cells (ECs), located at the interface between the circulating blood and the blood vessel, are exposed to shear stresses resulting from the tangential forces exerted by the flowing fluid on the vessel wall. The magnitude and pattern of the shear stress acting on ECs depend on blood flow, blood viscosity, and the vascular geometry, which varies with the location in the vascular tree. In regions of the vascular tree that have predilection for atherosclerotic lesions (e.g., branch points of large to medium arteries), the complex flow pattern is associated with low shear stresses that exhibit large spatial variations. In contrast, in the straight parts of the arterial tree, which are generally spared from atherosclerosis, blood flow is more laminar, and the high level of shear stress shows little spatial variations. Previous in vitro and in vivo findings have shown that ECs respond to shear stress in a magnitude- and flow pattern-dependent manner (3–5). ECs subjected to a long duration of laminar shear stress at the relatively high levels seen in the straight part of the arterial tree [i.e., on the order of 10–20 dynes (1 dyne = 10 μN)/cm2] have been found to have a lower rate of DNA synthesis than that under static condition (6). This shear-induced reduction of DNA synthesis, which indicates a decrease in cell proliferation, is not seen in ECs subjected to low shear stresses at 1–5 dynes/cm2 (6–8). The molecular mechanisms by which EC growth is regulated by the high level of sustained laminar shear stress seen in the lesion-resistant part of the arterial tree have not yet been clearly established. The elucidation of these mechanisms underlying the important medical problem of the regional predilection of atherogenesis requires an interdisciplinary approach combining engineering mechanics and molecular/cellular biology.
p53 is a transcription factor that is activated in various cell types in response to a variety of environmental stresses, including DNA-damaging agents, UV and ionizing irradiation, and hypoxia (9). In response to those stresses, p53 is accumulated as a result of an increased stability by phosphorylation at its N terminus (10). Several kinases, including casein kinases I and II, protein kinase A, cyclin-dependent kinase (cdk) 7, ataxia-telangioectasia mutated protein, and DNA-activated protein kinase, have been shown to phosphorylate p53 in vitro (10, 11). Recently, c-Jun N-terminal kinase (JNK) was shown to phosphorylate p53 in vitro with a concomitant increase in p53 half-life through a decrease in p53 ubiquitination (12, 13). JNK is known to mediate the signaling events in ECs in response to shear stress (14–16). Therefore, it is of interest to investigate the role of p53 in the shear-induced decrease in DNA synthesis and the effect of JNK on the shear regulation of p53.
An increase of p53 leads to the induction of growth arrest genes such as p21cip1 and the growth arrest and DNA damage inducible gene 45 (GADD45). GADD45 can be rapidly induced by various types of stresses, e.g., UV irradiation (17), and its overexpression in various types of tumor cells causes an inhibition of cell growth (18). p21cip1 causes an inhibition of cdks (e.g., cdk4 and cdk6) in many types of cells, including ECs (19, 20). A decrease in cdk activity causes retinoblastoma (Rb) hypophosphorylation, which occurs predominately in cells arrested at the G0/G1 phase. Hence, we tested the hypothesis that increases of p53 phosphorylation and expression level by shear stress lead to growth inhibition as a result of the induction of GADD45 and p21 and the ensuing Rb hypophosphorylation.
The current study shows that shear stress induces p53 in a magnitude- and time-dependent manner. Laminar shear stress at 12 dynes/cm2 causes a JNK-mediated phosphorylation of p53 and an increase of the p53 level. Laminar shear stress also causes increases in GADD45 and p21cip1 expression, a decrease of Rb phosphorylation, and a decrease in cell proliferation. This sequence of events provides a molecular mechanism by which long-term laminar shear stress inhibits EC proliferation.
Materials and Methods
Cell Culture.
The experiments were performed on bovine aortic endothelial cells (BAECs), which were chosen because of their high DNA transfection efficiency and established arterial lineage. BAECs before passage 10 were maintained in DMEM (GIBCO/BRL) supplemented with 10% FBS (GIBCO/BRL) in a humidified 5% CO2-95% air incubator at 37°C.
Shear Stress Experiment.
The parallel-plate flow chamber device (21) was used as an in vitro system to study the responses of cultured BAECs to laminar shear stress at the cellular and molecular levels. The basic design of the parallel-plate flow chamber is to allow a steady flow of fluid between two flat plates separated by a narrow gap created by using a gasket cutout. BAECs were cultured to confluence on the surface of one of the plates and exposed to the flow. The flow in the narrow gap between the two plates is laminar with a parabolic velocity profile. The wall shear stress on the surface of the plate (τwall) can be calculated as
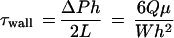 |
where Q is the volume flow rate, μ is the absolute fluid viscosity, and W, L, and h are the width, length, and gap height of the rectangular flow channel, respectively. ΔP is the pressure difference between the inlet and the outlet of the flow channel, and the desired ΔP level is attained by changing the relative vertical heights of two fluid reservoirs connected to the inlet and outlet of the flow chamber. The flow system was maintained at 37°C in a hood and equilibrated with 5% CO2-95% air.
Immunoblotting of p53, GADD45, p21, and Rb.
Confluent BAECs were sheared for various durations with various magnitudes of shear stress as indicated. Cytoplasmic proteins were extracted by using a lysis buffer containing 0.1% Triton X-100. After centrifugation, the pellet of the cell lysate containing the nuclear proteins was subjected to SDS/PAGE analysis followed by immunoblotting. p53 and Rb were detected by using antibodies obtained from PharMingen. GADD45 and P21cip1 were detected by using antibodies obtained from Santa Cruz Biotechnology.
p53 Phosphorylation Assay.
To determine whether JNK phosphorylates p53 in response to laminar shear stress, we performed JNK kinase activity assays by using GST-p53 as the substrate. Static or sheared BAECs were lysed in a lysis buffer containing 50 mM Hepes, pH 7.5, 0.15 M NaCl, 1 mM EDTA, 2.5 mM EGTA, 1% Triton X-100, 10 mM β-glycerophosphate, 1 mM NaF, 0.1 mM orthovanadate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM DTT, and 0.1 mM PMSF. JNK was immunoprecipitated with a polyclonal anti-JNK1 antibody (Santa Cruz) and protein A-Sepharose beads (Pharmacia Biotech). The kinase reaction was initiated by resuspending the immunoprecipitates in 30 μl of a kinase buffer [50 mM Hepes, pH 7.5/1 mM DTT/2.5 mM EGTA/10 mM MgCl2/20 mM ATP/10 μCi (γ-32P)ATP/0.1 mM orthovanadate/1 mM NaF/10 mM glycerophosphate], and 2 μg of GST-p53 was added as the substrate. After incubation at 30°C for 30 min, the reaction was terminated by the addition of 15 μl of ×3 SDS sample buffer containing 0.18 M Tris⋅HCl, pH 6.8, 30% (vol/vol) glycerol, 6% SDS, and 15% (vol/vol) β-mercaptoethanol. The phosphorylated GST-p53 was separated on a 10% SDS/PAGE and detected by autoradiography.
GADD45 Promoter Analysis.
GADD45 promoter/luciferase construct (22) and E6 plasmid were transfected into BAECs at 90% confluence by using the LipofectAmine method (GIBCO/BRL). The pSV-β-galactosidase (β-gal) plasmid, which contains a β-gal gene driven by the SV40 promoter and enhancer, was cotransfected to monitor the transfection efficiency. After transfection, the cells were either kept as static control or subjected to 12 dynes/cm2 of laminar shear stress for 24 h. The luciferase and β-gal activity assays were performed by following standard protocols.
Flow Cytometry for Cell Cycle Studies.
BAECs were pulse labeled with 10 μM BrdUrd during the last hour of shearing or static incubation. The cell isolation and labeling procedures were performed as described in the manufacturer's protocol (PharMingen). The BrdUrd-incorporated cells were detected by using FITC-conjugated anti-BrdUrd mAb. The total DNA content in the cell was stained by propidium iodine. The cells were analyzed by using the flow cytometer.
Results
Laminar Flow Causes a Magnitude- and Time-Dependent Increase in p53 Level in ECs.
BAECs were subjected to laminar shear stress of 0, 1.5, 3, 6, and 12 dynes/cm2 for 24 h and lysed for immunoblotting of p53. The application of laminar shear at 1.5 dynes/cm2 for 24 h did not increase the p53 level in ECs compared with that in static control kept for the same duration (Fig. 1A). An increase of shear stress to 3 dynes/cm2 increased the p53 level to 1.3 ± 0.2-fold of static control (P < 0.05). Further increases of shear stress to 6 and 12 dynes/cm2 increased the p53 induction to 1.6 ± 0.3- and 1.8 ± 0.6-fold, respectively. These results indicated that shear stress causes p53 with a threshold between 1.5 and 3 dynes/cm2. Beyond this threshold, the p53 level increased further as the shear stress was raised from 3 to 6 and 12 dynes/cm2. To investigate the time dependence of p53 induction by laminar shear stress, BAECs were subjected to a shear stress of 12 dynes/cm2 for various durations. As indicated in Fig. 1B, shearing for 1 h or longer caused increases in p53 in the BAECs. The increases were essentially the same between 2 and 24 h, indicating a sustained increase in p53 induction in response to a continued application of shear stress.
Figure 1.
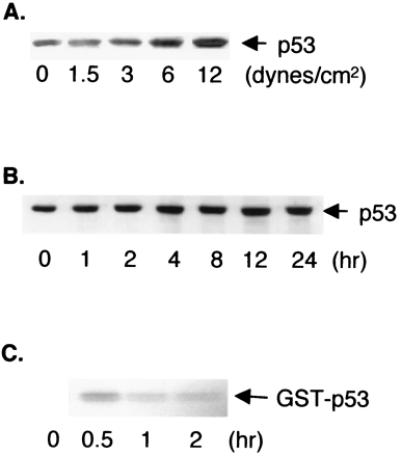
(A and B) Laminar shear stress increases the level of p53 in endothelial cells. In A, BAECs were subjected to laminar shear stress of 0, 1.5, 3, 6, and 12 dynes/cm2 for 24 h followed by immunoblotting by using an anti-p53 mAb. In B, BAECs were subjected to laminar shear stress at 12 dynes/cm2 for periods of time as indicated, followed by immunoblotting by using an anti-p53 mAb. (C) Laminar shear stress of 12 dynes/cm2 increases p53 phosphorylation by JNK. BAECs were subjected to laminar shear stress at 12 dynes/cm2 for periods of time as indicated or kept as static control represented by time 0. JNK was immunoprecipitated from BAEC lysates with an anti-JNK1 antibody, and the kinase activity of the isolated JNK was tested by using GST-p53 as a substrate in the presence of (γ-32P)ATP.
Laminar Flow Induces the Phosphorylation of p53 Through JNK.
It has been shown that JNK can serve as the upstream kinase for the phosphorylation of p53 to increase its stability in fibroblasts (12, 13). Our earlier studies demonstrated that laminar shear stress is a potent stimulus for JNK activation in ECs (16). Therefore, we investigated whether laminar shear-induced stabilization of p53 in ECs is because of its phosphorylation by JNK. JNK was immunoprecipitated from sheared and static BAEC lysates, and the kinase activity of the isolated JNK was assessed by using GST-p53 as the substrate. Fig. 1C shows that 30 min of laminar shear stress at 12 dynes/cm2 increased p53 phosphorylation by JNK and that this effect continued, although at a lesser level, throughout the 2-h period of study.
Laminar Flow Induces GADD45 and p21cip1 Expression.
Western blotting showed that the application of a laminar shear stress of 12 dynes/cm2 to BAECs for 24 h caused increases in the expressions of the growth arrest proteins GADD45 (Fig. 2A) and p21cip1 (Fig. 2B), as compared with the static control.
Figure 2.
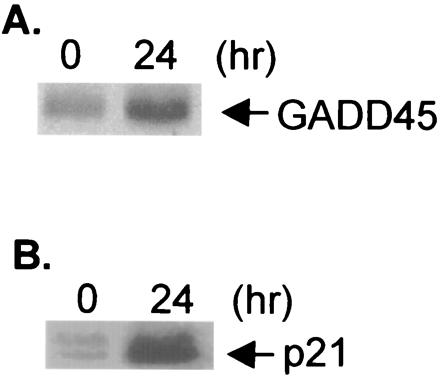
Laminar shear stress induces GADD45 and p21cip1 expression. Western blot analyses of the expression levels of GADD45 (A) and p21cip1 (B) were performed on BAECs subjected to laminar shear stress at 12 dynes/cm2 for 24 h.
Laminar Flow Induces GADD45 Promoter Activities Through p53.
To study the mechanism by which laminar shear stress regulates GADD45 gene expression, we used the reporter assay in which the activation of an appropriate promoter would lead to the expression of a linked reporter gene such as luciferase, which can be readily assessed by the use of a luminometer. BAECs were transfected with a wild-type (wt) GADD45 promoter linked to a luciferase reporter gene and subjected to laminar shear stress at 12 dynes/cm2 for 24 h. The results of the reporter assay (Fig. 3) show that laminar shear stress increased the activity of the wt GADD45 promoter by 12.4-fold, indicating that laminar shear stress causes the transcriptional expression of GADD45 through its wt promoter.
Figure 3.
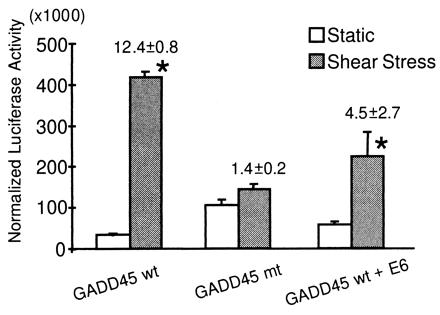
Laminar shear stress induces GADD45 promoter activity through the regulation of p53. BAECs were transfected with GADD45 promoters/luciferase plasmid. The transfections include the wt GADD45 promoter, a mutant GADD45 promoter, and wt GADD45 promoter together with an E6 expression construct. Transfected BAECs were subjected to laminar shear stress at 12 dynes/cm2 for 24 h. The cells were then lysed and assayed for luciferase activity. The GADD45 promoter activities were normalized by using the cotransfected β-gal expression plasmid. * denotes P < 0.05 compared with the corresponding controls kept under static condition. Note the absence of shear induction of the mutant promoter and the marked reduction in the shear-induced wt GADD promoter activity when cotransfected with an E6 expression construct.
The GADD45 promoter contains in its positions −204 to −190 a Wilms tumor suppressor gene 1 (WT1) binding site, the site on which p53 acts to regulate GADD45 promoter activity (22). We studied the effect of mutating this portion of the GADD45 promoter by replacing it with a TA-rich sequence. BAECs were transfected with such a mutant (mt) of the GADD45 promoter linked to a luciferase reporter gene. As shown in Fig. 3, laminar shear stress caused no significant induction of the mt GADD45 promoter, in contrast to the strong induction of wt GADD45 promoter.
To study further the involvement of p53 in the laminar shear-induced GADD45 expression, we used the E6 plasmid, which inhibits p53 activity by targeting it for degradation (23). The E6 plasmid was cotransfected with the luciferase reporter gene-linked wt GADD45 promoter into BAECs. As shown in Fig. 3, cotransfection of E6 plasmid markedly attenuated the laminar shear-induced wt GADD45 promoter activity (from 12.4- to 4.48-fold). This result indicates that p53 plays an important role in the induction of GADD45 expression by laminar shear stress.
Laminar Flow Induces Rb Hypophosphorylation.
Rb phosphorylation causes a decrease in p21cip1. Therefore, the increase of p21cip1 by laminar shear stress suggested this might be the result of a decrease in the phosphorylation level of Rb in the sheared cells. The results in Fig. 4 show that the application of laminar shear stress for 2 h or longer did cause Rb hypophosphorylation, which was sustained for at least 24 h. The degree of Rb phosphorylation is known to vary in the cell cycle, with the hypophosphorylated form being the major moiety of Rb in the G0/G1 phase. The Rb hypophosphorylation correlates well with our other findings of cell cycle arrest in response to laminar shear stress, i.e., increases in p53 (Fig. 1 A and B) and p21cip1 (Fig. 2B).
Figure 4.
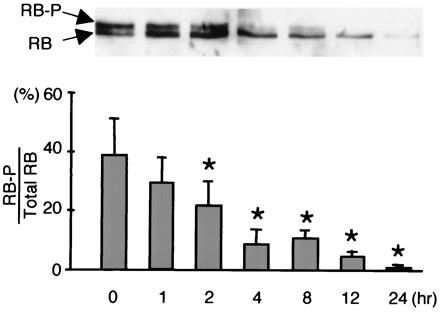
Laminar shear stress causes a decrease in the percentage of phosphorylated Rb in BAECs. Confluent BAECs were subjected to laminar shear stress at 12 dynes/cm2 for periods of time as indicated, or kept as static control represented by time 0. The amounts of hypo- and hyperphosphorylated Rb were determined by densitometry. “Rb-P” represents the hyperphosphroylated Rb and “Total Rb” is the sum of Rb-P and hypophosphorylated Rb. The bars represent the mean ± SD from three experiments. * denotes P < 0.05 compared with the static controls.
Laminar Flow Arrests EC Cell Cycle.
We used flow cytometry to analyze the effects of laminar shear stress on the distribution of cell population in different phases of the cell cycle. BAECs were either subjected to a laminar shear stress of 12 dynes/cm2 for 12, 24, or 36 h, or kept as static control for the same periods of time. Fig. 5 summarizes the results from four or more independent experiments. The results demonstrate that the decreases in the number of cells in S phase and the increases in G0/G1 under laminar shear stress are statistically significant for cells sheared for 24 and 36 h, compared with cells kept under static condition for the same lengths of time. After 12 h of shearing, only the decrease in S phase is significant, but not the increase in G0/G1. The decrease in the percentage of cells in S phase suggests that laminar shear stress may prevent ECs from entering the S phase. The increase in the percentage of cells in G0/G1 phase by shear stress suggests a cell cycle arrest in G0/G1.
Figure 5.
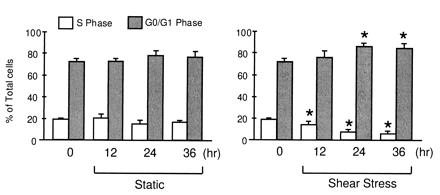
Modulations of BAEC cell cycle by laminar shear stress. Open bars represent S phase; shaded bars denote G0/G1 phase. The data plotted are mean ± SD from four independent experiments. * denotes P < 0.05 compared with the corresponding controls kept under static condition for the same lengths of time. (Left) Results on ECs under static condition. (Right) Results on ECs under laminar shear stress of 12 dynes/cm2.
Discussion
ECs in the straight parts of the arterial tree are constantly exposed to high levels of shear stress. In contrast, ECs at the arterial branches are exposed to disturbed flow with lower shear stress and more spatial variations. Static EC cultures in vitro, which are not exposed to shear stress, have higher turnover rates than the ECs exposed to laminar shear stress (24). In line with these findings, DNA synthesis rate in cultured ECs exposed to disturbed flow in a step flow channel (25) is highest in the region of flow reattachment, where the shear stress is near zero with large spatial variations. The exposure of ECs to laminar flow at high shear stress inhibits their DNA synthesis (24). Although previous findings suggested a shear stress magnitude-dependent inhibition of DNA synthesis in ECs under laminar shear stress, the underlying mechanism is still unclear. In this study, we demonstrated the stress magnitude-dependent increase of p53 in ECs (Fig. 1). The threshold of shear stress for inducing p53 is between 1.5 and 3 dynes/cm2. At the branch points of arterial vessels, the shear stress can be lower than 1 dyne/cm2 because of secondary flow and flow reattachment. In the straight regions, the average shear stress is well above 3 dynes/cm2, and the flow pattern is laminar with little spatial variation.
The tumor suppressor protein p53, acting as a transcription factor, is believed to mediate a number of cellular functions, including cell cycle arrest and apoptosis, in response to various environmental stresses. UV irradiation, which causes EC apoptosis (26), induces a strong transient increase of p53. In contrast, the application of laminar shear stress to ECs induced a moderate but sustained increase in p53 in the present study (Fig. 1B) and did not cause apoptosis in our previous study (14). Sustained laminar shear stress, in addition to sparing EC from apoptosis, is capable of preventing the EC apoptosis induced by cytokines (e.g., tumor necrosis factor-α), oxidative stress, serum starvation, or cytoskeletal disruption (14, 27–29). The different phenotypic responses of ECs exposed to UV irradiation vs. laminar shear stress suggest that the time course and intensity of the p53 response may be critical in determining cell fates. It appears that the sustained and moderate level of p53 elevation induced by laminar shear stress causes cell cycle arrest, enhances DNA repair, and serves a protective function against excessive cell proliferation and cell death.
In investigating the mechanism of the increase in p53 level in response to laminar shear stress, we found that the shear-induced JNK activity can phosphorylate p53 in vitro. The time course of the p53 phosphorylation by JNK, with a peak at 30 min, correlates well with that of the JNK response to shear stress (16). Because the increase in p53 level persists for 24 h after shearing, other shear-induced kinases, such as ataxia-telangioectasia mutated protein and DNA-activated protein kinase, may contribute to this sustained increase, and it appears that JNK plays a role in mediating the increase of p53 mainly in the early phase.
The involvement of GADD45 in cell cycle arrest at G0/G1 or G2/M phase has been suggested in other systems (30, 31). The GADD45 promoter contains no p53-binding site, and the p53 responsiveness is located in a GC-rich motif containing a WT1 site (22). These results indicate that p53 regulates the GADD45 promoter through protein–protein interaction (22). In our experiments, laminar shear stress caused a 12-fold increase in GADD45 promoter activity. This shear-induced GADD45 promoter activity was markedly attenuated by inhibition of the p53 transcription activity with a viral protein E6 and was essentially abolished by mutation of the WT1 site. Hence, our results indicate that shear-induced GADD45 transcription is mediated, at least in part, through the interaction of p53 with WT1.
P21cip1 regulates the cell cycle by binding to cdk/cyclin complex and acting as a cdk inhibitor. The phosphorylation of Rb by cdk is the critical step for liberating cells from growth suppression (quiescent G0/G1 phase) to enter the cell cycle (S phase). Our findings indicate that laminar shear stress induces p21cip1 expression, which may then inhibit the cdk-mediated Rb phosphorylation. Our findings on the shear-induced Rb hypophosphorylation and p21 expression are in agreement with those in the paper by Akimoto et al. (32), which we discovered when revising our manuscript. Our study has provided the additional findings that p53 (a p21 up-stream regulator) and GADD45 (another cell growth arrest gene) are also involved in the shear-induced EC arrest.
In conclusion, our findings suggest that the EC cell cycle arrest induced by long-term laminar shear stress is mediated, at least in part, through the p53 pathway. Laminar shear stress increases the p53 level and induces prolonged expressions of GADD45 and p21cip1 in ECs. The inhibition of cdk activity resulting from the augmentation of p21cip1 causes Rb hypophosphorylation, which leads to the EC cell cycle arrest at G0/G1 phase. These results provide insights into the molecular mechanisms by which laminar flow inhibits EC proliferation to serve a protective role against atherogenesis in the straight parts of the arterial tree. Coupled with our previous study on the enhancement of EC proliferation at reattachment regions that simulate the flow pattern at arterial branch points (25), these investigations serve to link the mechanics of blood flow to molecular signaling in endothelial cells and their functional behavior in health and disease.
Acknowledgments
We thank Dr. Albert Fornace of the National Cancer Institute, National Institutes of Health, Bethesda, MD, for providing the GADD45 promoter. We also thank Dr. Shi Huang of the Burnham Institute, La Jolla, CA for providing the GST-p53 construct for p53 phosphorylation analysis. This work is supported by a gift from Dr. Shi H. Huang of the Chinfong Group, Taipei, and by grants HL-19454, HL-43026, HL-64382 (S.C.), and HL-56707, HL-60789 (J.S.) from the National Heart, Lung, and Blood Institute.
Abbreviations
- cdk
cyclin-dependent kinase
- EC
endothelial cell
- JNK
c-Jun N-terminal kinase
- PCNA
proliferating cell nuclear antigen
- Rb
retinoblastoma
- wt
wild type
- WT1
Wilms tumor suppressor gene 1
- β-gal
β-galactosidase
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170282597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170282597
References
- 1.Rossitti S. Arq Neuro-Psiquiatr. 1995;53:333–341. doi: 10.1590/s0004-282x1995000200028. [DOI] [PubMed] [Google Scholar]
- 2.Langille B L. Can J Physiol Pharmacol. 1996;74:834–841. [PubMed] [Google Scholar]
- 3.Noris M, Morigi M, Donadelli R, Aiello S, Foppolo M, Todeschini M, Orisio S, Remuzzi G, Remuzzi A. Circ Res. 1995;76:536–543. doi: 10.1161/01.res.76.4.536. [DOI] [PubMed] [Google Scholar]
- 4.Malek A M, Izumo S. J Biomech. 1995;28:1515–1528. doi: 10.1016/0021-9290(95)00099-2. [DOI] [PubMed] [Google Scholar]
- 5.Boegehold M A. Am J Physiol. 1996;271:H387–395. doi: 10.1152/ajpheart.1996.271.2.H387. [DOI] [PubMed] [Google Scholar]
- 6.Nerem R M. Monogr Atheroscler. 1990;15:117–124. [PubMed] [Google Scholar]
- 7.Davies P F, Remuzzi A, Gordon E J, Dewey C J, Gimbrone M J. Proc Natl Acad Sci USA. 1986;83:2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dewey C J, Bussolari S R, Gimbrone M J, Davies P F. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 9.Levine A J. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 10.Steegenga W T, van, der, Eb Aj, Jochemsen A G. J Mol Biol. 1996;263:103–113. doi: 10.1006/jmbi.1996.0560. [DOI] [PubMed] [Google Scholar]
- 11.Meek D W, Campbell L E, Jardine L J, Knippschild U, McKendrick L, Milne D M. Biochem Soc Trans. 1997;25:416–419. doi: 10.1042/bst0250416. [DOI] [PubMed] [Google Scholar]
- 12.Adler V, Pincus M R, Minamoto T, Fuchs S Y, Bluth M J, Brandt R P, Friedman F K, Robinson R C, Chen J M, Wang X W, et al. Proc Natl Acad Sci USA. 1997;94:1686–1691. doi: 10.1073/pnas.94.5.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs S Y, Adler V, Buschmann T, Yin Z, Wu X, Jones S N, Ronai Z. Genes Dev. 1998;12:2658–2663. doi: 10.1101/gad.12.17.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y L, Li S, Shyy J Y, Chien S. Am J Physiol. 1999;277:H1593–H1599. doi: 10.1152/ajpheart.1999.277.4.H1593. [DOI] [PubMed] [Google Scholar]
- 15.Jo H, Sipos K, Go Y M, Law P, Rong J, McDonald J M. J Biol Chem. 1997;272:1395–1401. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- 16.Li Y S, Shyy J Y, Li S, Lee J, Su B, Karin M, Chien S. Mol Cell Biol. 1996;16:5947–5954. doi: 10.1128/mcb.16.11.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papathanasiou M A, Fornace A J. Cancer Treat Res. 1991;57:13–36. doi: 10.1007/978-1-4615-3872-1_2. [DOI] [PubMed] [Google Scholar]
- 18.Zhan Q, Fan S, Bae I, Guillouf C, Liebermann D A, O'Connor P M, Fornace A J. Oncogene. 1994;9:3743–3751. [PubMed] [Google Scholar]
- 19.Hsieh T C, Juan G, Darzynkiewicz Z, Wu J M. Cancer Res. 1999;59:2596–2601. [PubMed] [Google Scholar]
- 20.Schonthal A H, Hwang J J, Stevenson D, Trousdale M D. Exp Eye Res. 1999;68:531–539. doi: 10.1006/exer.1998.0634. [DOI] [PubMed] [Google Scholar]
- 21.Frangos J A, Eskin S G, McIntire L V, Ives C L. Science. 1985;227:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 22.Zhan Q, Chen I T, Antinore M J, Fornace A J. Mol Cell Biol. 1998;18:2768–2778. doi: 10.1128/mcb.18.5.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etscheid B G, Foster S A, Galloway D A. Virology. 1994;205:583–585. doi: 10.1006/viro.1994.1684. [DOI] [PubMed] [Google Scholar]
- 24.Levesque M J, Nerem R M, Sprague E A. Biomaterials. 1990;11:702–707. doi: 10.1016/0142-9612(90)90031-k. [DOI] [PubMed] [Google Scholar]
- 25.Chiu J J, Wang D L, Chien S, Skalak R, Usami S. J Biomech Eng. 1998;120:2–8. doi: 10.1115/1.2834303. [DOI] [PubMed] [Google Scholar]
- 26.Suschek C V, Krischel V, Bruch G D, Berendji D, Krutmann J, Kroncke K D, Kolb B V. J Biol Chem. 1999;274:6130–6137. doi: 10.1074/jbc.274.10.6130. [DOI] [PubMed] [Google Scholar]
- 27.Dimmeler S, Haendeler J, Rippmann V, Nehls M, Zeiher A M. FEBS Lett. 1996;399:71–74. doi: 10.1016/s0014-5793(96)01289-6. [DOI] [PubMed] [Google Scholar]
- 28.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher A M. Circ Res. 1998;83:334–341. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 29.Dimmeler S, Hermann C, Galle J, Zeiher A M. Arterioscler Thromb Vasc Biol. 1999;19:656–664. doi: 10.1161/01.atv.19.3.656. [DOI] [PubMed] [Google Scholar]
- 30.Smith M L, Chen I T, Zhan Q, Bae I, Chen C Y, Gilmer T M, Kastan M B, O'Connor P M, Fornace A J. Science. 1994;266:1376–1380. doi: 10.1126/science.7973727. [DOI] [PubMed] [Google Scholar]
- 31.Wang X W, Zhan Q, Coursen J D, Khan M A, Kontny H U, Yu L, Hollander M C, O'Connor P M, Fornace A J, Harris C C. Proc Natl Acad Sci USA. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akimoto S, Mitsumata M, Sasaguri T, Yoshida Y. Cir Res. 2000;86:185–190. doi: 10.1161/01.res.86.2.185. [DOI] [PubMed] [Google Scholar]