

Abstract
The evolution of proteins for novel functions involves point mutations and recombinations of domains or structural segments. Mimicking this process by rational design in vitro is still a major challenge. The present report demonstrates that the active site of the enzyme glutathione transferase (GST) A1–1 can be tailored for high catalytic efficiency with alkenals. The result is a >3,000-fold change in substrate selectivity involving a noteworthy change in preferred catalyzed reaction from aromatic nucleophilic substitution to Michael addition. The hydrophobic substrate binding pocket of GST A1–1 is formed by three structural modules, which were redesigned sequentially with four point mutations and the exchange of a helical segment. The substitutions were made to mimic first-sphere interactions with a substrate in GST A4–4, which naturally has high activity with alkenals. These substrates are toxic lipid peroxidation products of pathophysiological significance, and glutathione conjugation is a route of their inactivation. The final product of the sequential redesign of GST A1–1, mutant GIMFhelix, had a 300-fold increase in catalytic efficiency with nonenal and a >10 times decreased activity with 1-chloro-2,4-dinitrobenzene. In absolute values, GIMFhelix is more efficient than wild-type GST A4–4 with some alkenal substrates, with a kcat/Km value of 1.5 ± 0.1 106 M-1⋅s−1 for nonenal. The pKa value of the active-site Tyr-9 of GIMFhelix is 7.3 ± 0.1, approaching the unusually low value of GST A4–4. Thus, rational redesign of the active-site region of an enzyme may be sufficient for the generation of efficient catalysts with altered chemical mechanism and novel selectivity.
Keywords: hydroxyalkenal, protein redesign, sequential design
In nature, evolution operates by selection for fitness among biological or chemical species. In vitro, similar evolution can be afforded by combinatorial protein chemistry in conjunction with proper tests for fitness (1). The family of glutathione transferases (GSTs) is an example of proteins where currently existing isoenzymes bear evidence of natural combinatorics at all structural levels: primary, secondary, tertiary, and quaternary. Variations in primary structures are obvious among isoenzymes, and acquisition or loss of a secondary structure element such as a helix is noted when GSTs of different classes are compared (2). At the tertiary structural level, the glutathione-binding domain has the same fold as thioredoxin and a domain of glutathione peroxidase, suggesting that this supersecondary structure is a building block recruited for different purposes in molecular evolution (2, 3). Finally, GST subunits form binary combinations at the level of quaternary structure such that homodimers and heterodimers naturally occur (4). However, it is not obvious how this understanding of protein structure can be exploited for the engineering of enzymes with novel functions.
Glutathione-linked reactions, catalyzed by GSTs, play a central role in cellular detoxication of electrophilic toxic and carcinogenic molecules, in particular the primary and secondary products of oxidative stress (5–7). Available evidence strongly indicates that GSTs provide protection against development of cancer (8) and other degenerative diseases, e.g., atherosclerosis (9), cataract (10), and Parkinson's disease (11). GST A4–4 appears to play a particularly prominent role in the protection against degenerative processes, because of its high catalytic activity with alkenals (12, 13) that are formed by radical reactions and oxidative damage to lipids and other biomolecules (14).
As a superfamily of enzymes GSTs have evolved the ability to interact with literally thousands of chemical structures of endogenous and xenobiotic origins (6, 15). Even if the catalytic efficiency is low with the majority of the ligands that can be bound to a particular GST, high activity with a given substrate can be acquired by proper interplay of structural redesign and selection methods.
With biologically relevant substrates, such as alkenals (12, 14), the catalytic efficiencies of some GSTs (e.g., GST A4–4) are highly evolved and differ from those of other isoenzymes by several orders of magnitude. GSTs thus may be regarded as separate enzymes that are using the same cofactor (glutathione) in analogy to, e.g., the multiple pyridine-nucleotide-linked dehydrogenases. The soluble GSTs have the same fold of the polypeptide chain and the same overall structure with an essentially conserved glutathione binding site (2, 16), suggesting that modifying the structural elements that form the substrate-binding cavity would be sufficient to generate a new GST with altered substrate specificity. The determinants of the substrate selectivity are believed to involve a limited set of amino acid residues in three distinct structural modules: the β1-α1 loop following the first β-strand, the C-terminal one-third of the α4-helix, and the C terminus (Fig. 1).
Figure 1.

The three hydrophobic substrate-binding modules of human GST A1–1 (2) and human GST A4–4 (17): the β1-α1 loop (residues 9–16), the C-terminal part of the α4 helix (residues 104–111), and the C terminus of the protein (residues 208–222, where 210–220 form the α9 helix). GST A4–4 residues considered important for proper first-sphere interactions with alkenal substrates and installed into corresponding positions in GST A1–1 are boxed. Residue numbers are indicated above the residues at each end.
The availability of three-dimensional structures and methods for genetic engineering has made possible rational modifications of functional properties of proteins. The basic assumption is that the amino acid side chains contacting the substrate or other ligands in a binding site determine the affinity of a bound molecule and the chemical transformation that it may undergo. Comparisons of primary structures of homologous proteins also have given valuable clues to the identification of structural determinants of function. For example, the coenzyme preference of pyridine-nucleotide-dependent oxidoreductases has been reversed (18, 19), the substrate specificity of trypsin has been transformed to mimic that of chymotrypsin (20), chemical reactions involving fatty acids (21) and steroids (22) have been changed, and aspartate aminotransferase has been altered into a tyrosine aminotransferase (23). Following the original work from Bender's (24) and Koshland's (25) laboratories the active-site serine hydroxyl group of subtilisin has been chemically modified to a selenol, which confers peroxidase activity to the hydrolase (26).
To further explore the rational approach to redesign of the catalytic function of enzymes, an attempt was made to install the high alkenal activity of GST A4–4 into the low-activity enzyme GST A1–1. The amino acid sequences of GST A1–1 and GST A4–4 are 53% identical and their protein folds are essentially the same, but their substrate selectivities are distinctly different (13). A successful redesign would alter the catalytic selectivity of GST A1–1 from aromatic substitution reactions to Michael additions, characterizing GST A4–4 (Fig. 2). Mutations in the three substrate-binding modules of GST A1–1 (Fig. 3) were introduced cumulatively, and a chimeric protein was constructed in which a C-terminal segment of GST A1–1 had been replaced with that of GST A4–4 (Table 1). The ultimate mutant, GIMFhelix, is catalytically more efficient with hexenal and nonenal than the naturally evolved GST A4–4.
Figure 2.
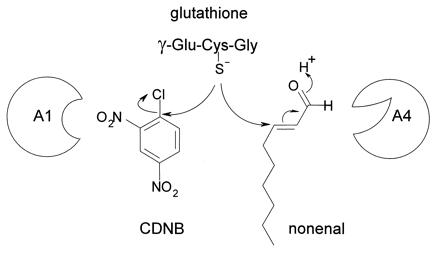
Two distinct chemical reactions efficiently catalyzed by GST A1–1 and GST A4–4. The reaction mechanisms differ; GST A1–1 favors an aromatic substitution reaction by which the sulfur of glutathione (γ-Glu-Cys-Gly) replaces the chlorine atom of CDNB. GST A4–4 has the highest activity in the Michael addition reaction where glutathione is added to the activated double bond of an alkenal. One subunit of each enzyme is shown (A1 and A4).
Figure 3.
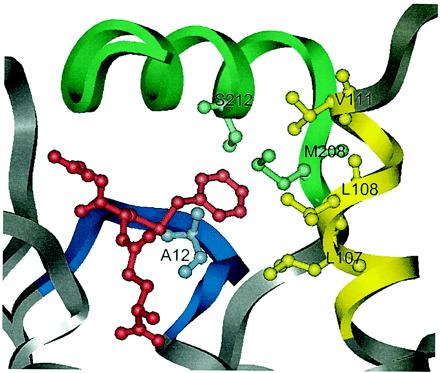
Close view of the active site of GST A1–1 (2). The ligand, S-benzylglutathione, which defines the position of the active site, is colored red. The following mutations have been introduced in mutant GIMFhelix: Ala-12–Gly in the β1-α1 loop (blue) and Leu-107–Ile, Leu-108–Met, and Val-111–Phe in the C-terminal part of α4-helix (yellow). The C terminus (green) of GST A1–1 has been replaced with the corresponding residues of GST A4–4. This substitution altered 10 amino acid residues and included the mutations Ser-212–Tyr and Met-208–Pro, believed to be important for the change of the mechanism to a Michael addition.
Table 1.
GST A1-1 mutants constructed by site-directed mutagenesis
| Enzyme | Mutations |
|---|---|
| mut1 | S212Y |
| mut2 | A12G, S212Y |
| mut3 | A12G, M208P, S212Y |
| Ghelix | A12G, helixA4 (residues 208–222) |
| GIMFhelix | A12G, L107I, L108M, V111F, helixA4 |
Methods
Mutagenesis and Heterologous Protein Expression.
Clones of wild-type human GST A1–1 (27) and GST A4–4 (13) and their heterologous expression in Escherichia coli have been described. Mut1 (Table 1) was constructed with the GST A1–1 clone as template, and the other mutations were added sequentially by use of custom synthesized oligonucleotide primers and PCR with Taq DNA polymerase, or by inverted PCR with Pfu DNA polymerase. The coding sequences were subjected to sequence analysis to verify the mutations and subcloned into the expression vector pET-21a(+) (Novagen) by using NdeI and SalI restriction sites. The GST A1–1 mutants were expressed in E. coli BL-21 and purified by methods previously described (28). All proteins used were pure as judged from SDS/PAGE (29). The following extinction coefficients were used to determine the protein subunit concentrations: GST A1–1, ɛ280 = 24,700 M-1⋅cm-1 (30); GST A4–4, ɛ280 = 15,930 M-1⋅cm-1; mut1 and mut2, ɛ280 = 19,770 M-1⋅cm-1; mut3 and Ghelix, ɛ278 = 22,400 M-1⋅cm-1; and GIMFhelix: ɛ276 = 22,800 M-1⋅cm-1. All of the extinction coefficients but that for GST A1–1 were calculated as described (31).
Kinetic Measurements.
Kinetic studies were conducted spectrophotometrically in 0.1 M sodium phosphate at pH 6.5 and 30°C (32, 33). The kinetic constants, kcat, Km, and kcat/Km (expressed per subunit) were obtained by nonlinear regression analysis using the following extinction coefficients for 1-chloro-2,4-dinitrobenzene (CDNB), ɛ340 = 9.6 mM-1⋅cm-1 (32); hexenal, ɛ224 = 19.5 mM-1⋅cm-1 (13); nonenal, ɛ225 = 19.22 mM-1⋅cm-1 (13), and hydroxyalkenals, ɛ224 = 13.75 mM-1⋅cm-1 (34). trans-2-Hexenal and trans-2-nonenal were purchased from Aldrich. 4-Hydroxy-trans-2-hexenal and 4-hydroxy-trans-2-nonenal were kind gifts of Hermann Esterbauer, University of Graz, Graz, Austria.
Determination of Active-Site Tyrosine pKa.
The concentration of deprotonated Tyr-9 in the active site was determined as a function of pH by use of difference UV spectroscopy, and the pKa value was estimated by fitting a pH titration curve to the data as described (28, 35).
Results
Substrates Used for the Characterization of Mutants.
CDNB, hexenal, and nonenal were chosen as representative substrates for monitoring the progress in the sequential redesign of GST A1–1. CDNB and nonenal afford high catalytic efficiencies in the glutathione conjugation reactions catalyzed by GST A1–1 and GST A4–4, respectively. Hexenal was chosen as an alternative alkenal substrate with a shorter chain length. Ghelix and GIMFhelix also were compared with GST A4–4 by using the pathophysiologically important lipid peroxidation products (36) 4-hydroxyhexenal (HHE) and 4-hydroxynonenal (HNE).
Kinetic Characterization of Point Mutants.
The consecutive point mutants, mut1, mut2, and mut3 (see Table 1), all have kcatnonenal values elevated 5–6 times compared with GST A1–1 (Table 2). The Kmnonenal values were moderately increased (up to 2-fold), giving catalytic efficiencies (kcat/Km) 3–5 times higher than for GST A1–1. With hexenal kcat/Km increases gradually for each added mutation almost in the same manner as with nonenal.
Table 2.
Kinetic parameters of human GST A1-1 and its mutated variants compared to those of human GST A4-4
| Enzyme | CDNB
|
Hexenal
|
Nonenal
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Km, mM | kcat, s−1 | kcat/Km, mM−1⋅s−1 | Km, mM | kcat, s−1 | kcat/Km, mM−1⋅s−1 | Km, mM | kcat, s−1 | kcat/Km, mM−1⋅s−1 | |
| A1-1 | 0.56 ± 0.04* | 88 ± 3* | 160* | 0.18 ± 0.02 | 0.22 ± 0.02 | 1.2 ± 0.1 | 0.025 ± 0.004 | 0.13 ± 0.01 | 5.0 ± 0.6 |
| mut1 | 3.2 ± 1.9 | 62 ± 25 | 19 ± 4 | ND | ND | 0.059 ± 0.008 | 0.051 ± 0.012 | 0.70 ± 0.09 | 14 ± 2 |
| mut2 | 4.2 ± 1.3 | 99 ± 22 | 24 ± 2 | 0.90 ± 0.36 | 2.5 ± 0.9 | 2.7 ± 0.1 | 0.035 ± 0.004 | 0.79 ± 0.03 | 23 ± 2 |
| mut3 | 3.7 ± 1.7 | 29 ± 9 | 7.9 ± 1.1 | ND | ND | 4.7 ± 0.2 | 0.035 ± 0.008 | 0.60 ± 0.05 | 17 ± 3 |
| Ghelix | 12 ± 9 | 81 ± 55 | 7.0 ± 0.7 | 0.48 ± 0.10 | 3.2 ± 0.7 | 6.8 ± 0.2 | 0.076 ± 0.012 | 4.3 ± 0.4 | 57 ± 5 |
| GIMFhelix | 3.4 ± 2.2 | 48 ± 22 | 14 ± 3 | 0.43 ± 0.09 | 48 ± 9 | 113 ± 4 | 0.014 ± 0.002 | 21 ± 1 | 1,520 ± 120 |
| A4-4 | 5.8 ± 1.4† | 46 ± 9† | 7.9† | 0.36 ± 0.23 | 13 ± 7 | 37 ± 4 | 0.180 ± 0.050 | 89 ± 18 | 485 ± 43 |
Substrate-saturation curves were measured by using CDNB (0.2–1.8 mM) or alkenals (0.01–0.10 mM) as the varied substrate at constant concentration of glutathione (5 mM and 0.5 mM, respectively). Higher substrate concentrations could not be used for reasons of limited solubility and high absorbance. However, kcat/Km values could still be determined with adequate precision by fitting v = (kcat/Km) [S] [E]o/(1 + [S]/Km) to the experimental data. ND, not possible to determine.
From ref. 30.
From ref. 13.
All mutations of GST A1–1 caused a decrease in the kcat/Km for CDNB by an order of magnitude (Table 2). The major effect is on KmCDNB, which is increased from the GST A1–1 value of 0.56 mM and approaching the GST A4–4 value of 5.8 mM.
Kinetic Characterization of the Chimeric Proteins.
Replacement of the C terminus (residues 208–222) of GST A1–1 with that from GST A4–4 (to give the chimeric mutant Ghelix) was a breakthrough in the sequential redesign, by which the kcat/Km values for alkenals rose toward those of GST A4–4 (Table 2). With the additional three mutations Leu-107–Ile, Leu-108–Met, and Val-111–Phe, (providing mutant GIMFhelix) the kcat/Km values for hexenal and nonenal finally exceeded the values of GST A4–4, by a factor of approximately 3. In comparison with the original enzyme, GST A1–1, the high catalytic efficiency is primarily caused by an increase in the kcat value, which is raised 200 and 160 times for hexenal and nonenal, respectively. It is noteworthy that the Kmhexenal value is almost the same for GIMFhelix as for GST A4–4, whereas Kmnonenal for GIMFhelix is 1 order of magnitude lower than the GST A4–4 value.
Catalytic Efficiencies with HHE and HNE.
GST A4–4 has a higher catalytic efficiency with 4-hydroxyalkenals than with their nonhydroxylated analogs (Fig. 4). The difference is most pronounced with HNE, for which the effect is caused by a Km value that is lower than for nonenal by a factor of approximately 5. Neither Ghelix nor GIMFhelix shows any major differences between HNE and nonenal in the measured kinetic parameters (Table 2 and Fig. 4). Compared with the redesigned GST A1–1 variants, GST A4–4 has a kcatHNE value that is higher by 1 order of magnitude.
Figure 4.
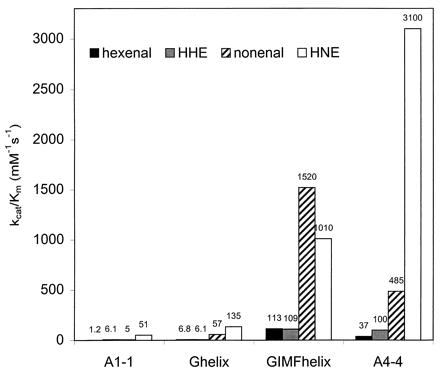
Substrate selectivity profiles expressed as catalytic efficiency, kcat/Km, of mutants Ghelix and GIMFhelix as well as of wild-type GST A1–1 and GST A4–4, with the following substrates: hexenal (black), hydroxyhexenal, HHE (gray), nonenal (hatched), and HNE (white). The kcat/Km values for GST A4–4 obtained with HHE and HNE were determined previously (13). The kinetic parameters for hexenal and nonenal are from Table 2, and for HHE and HNE as follows: GST A1–1, KmHNE = 0.047 ± 0.002 mM, kcatHNE = 2.4 ± 0.05 s-1; Ghelix, KmHNE = 0.047 ± 0.017 mM, kcatHNE = 6.3 ± 1.3 s-1; GIMFhelix, KmHHE = 0.39 ± 0.07 mM, kcatHHE = 42.9 ± 6.4 s-1, KmHNE = 0.011 ± 0.003 mM, kcatHNE = 11.6 ± 0.7 s-1; and GST A4–4 (13), KmHHE = 0.23 ± 0.05 mM, kcatHHE = 23 ± 3 s-1, KmHNE = 0.037 ± 0.004 mM, kcatHNE = 113 ± 4 s-1.
Effects of Mutations on the Ionization of the Active Site Tyr-9.
Tyr-9 has been identified as an important active-site residue for the class Alpha GSTs (2, 37). It is characterized by a low pKa value of 8.1 ± 0.1 in GST A1–1 (35). However, the pKa of Tyr-9 in GST A4–4 is even lower 6.7 ± 0.2 (I. Hubatsch and B.M., unpublished work). The pKa value of Ghelix was determined as 8.0 ± 0.2, whereas a significantly lower value of 7.3 ± 0.1 was obtained for GIMFhelix, showing that the redesigned GST A1–1 had gained resemblance to GST A4–4.
Discussion
Rationale for the Mutations.
The first attempt to elucidate the structural basis for the high activity of GST A4–4 with alkenals was made (38) by construction of chimeras of rat GST A1–1 and rat GST A4–4 (earlier called GST 1–1 and GST 8–8). It was shown that a glycine residue in position 12 was required to afford high catalytic efficiency with alkenals. However, introduction of this amino acid residue into wild-type rat GST A1–1 did not result in elevated alkenal activity, implying that additional residues are crucial for catalysis of the Michael addition.
The crystal structure of human GST A4–4 (17) revealed that Tyr-212, in the C-terminal helix of GST A4–4, might polarize the carbonyl group of the alkenal substrate and thereby facilitate the Michael addition of glutathione. This mechanistic feature was supported by mutagenesis of Tyr-212 (17). Glycine in position 12 is required for the Tyr to adopt the position that assists catalysis. Any other residue would have a β-carbon sterically interfering with Tyr-212 (17). Thus, the Ala-12–Gly and Ser-212–Tyr mutations were introduced in the redesign of GST A1–1. Proline in position 208 is highly conserved in GSTs with alkenal activity, whereas the corresponding residue in GST A1–1 is methionine. A comparison of the crystal structures of GST A1–1 (2) and GST A4–4 (17) showed that Met-208 also might interfere with the tyrosine introduced in position 212. Thus, residue 208 also was targeted.
The positioning and packing of amino acid residues create very different shapes of the substrate-binding pockets in the two enzymes. Seven residues line the alkenal-binding groove in GST A4–4: Gly-14, Ile-107, Met-108, Phe-111, Tyr-212, Val-216, and Tyr-217. Additionally, Val-213 may be of importance as a second-sphere component, forming a part of the buried hydrophobic core supporting the core structure of the binding site (17). Of these, four residues are conserved exclusively in GSTs with high alkenal activity: Phe-111, Tyr-212, Val-213, and Val-216. This was the rationale for changing these residues.
Arg-221 and Pro-222 are disordered in the GST A4–4 structure and the location of their side chains could not be determined (17). However, in the structure of GST A1–1 complexed with S-benzylglutathione the corresponding residues, Arg-221 and Phe-222, are well defined (2), and the aromatic side chain of Phe-222 is in a position likely to hinder the binding of long chain alkenal substrate. The C-terminal helix (residues 210–220) of GST A4–4 seems to be more stable and have a somewhat different location relative to the active site (17) in comparison to the flexible C terminus of GST A1–1 (2, 39). Only three of 11 residues in the C-terminal helix are conserved between GST A1–1 and GST A4–4 and the sequence difference might account for the distinct properties of the helices. Therefore, the whole C terminus was exchanged, rather than only making substitutions of residues 208, 212, 213, 216, and 222.
Mutant with the Highest Alkenal Activity.
GIMFhelix was created by grafting the GST A4–4 residues 12, 107, 108, and 111 and the C-terminal peptide (residues 208–222) into the structure of GST A1–1. The amino acid sequences of GST A1–1 and GST A4–4 are highly divergent and GIMFhelix, like the other mutants, is thus predominantly a GST A1–1 molecule (with 14 aa, i.e., 6%, substituted). Based on the specificity constant, kcat/Km, mutant GIMFhelix is 3-fold more efficient than GST A4–4 with the substrates nonenal and hexenal (Table 2 and Fig. 4). In relation to the parental GST A1–1, mutant GIMFhelix is >300-fold more efficient with nonenal. The increased catalytic efficiency with alkenals is selective, because the xenobiotic substrate CDNB displayed a >10 times decreased activity with the mutant in comparison with GST A1–1 (Table 2). The ratio of the activities for the Michael addition and the aromatic substitution consequently increased >3,000 fold, a significant measure of the altered catalytic profile. The pKa value of the active-site Tyr-9 of mutant GIMFhelix (7.3) is shifted close to that of GST A4–4 (6.7), further indicating that the redesign has been successful.
Activities Measured with the Physiologically Relevant HHE and HNE Substrates.
GIMFhelix and GST A4–4 have essentially the same catalytic efficiency with HHE, although both Km and kcat values are higher for GIMFhelix (Fig. 4). With HNE GST A4–4 displays a higher catalytic efficiency, because of a higher kcat value. The alkenals used as substrates are all in the trans configuration, but the 4-hydroxyalkenals have a chiral center at C4, which gives a mixture of enantiomers. Modeling suggests that there is room for the hydroxyl group on C4 in both configurations in the binding site of GST A4–4 (17), but in GIMFhelix this might not be the case.
A Successful Rational Redesign.
The > 3,000-fold alteration of the substrate selectivity of GST A1–1, accompanied by a transmutation of the favored catalyzed reaction from aromatic substitution to Michael addition, was achieved by a limited number of rationally chosen active-site mutations. With alkenals GIMFhelix matches the most efficient isoenzyme known, GST A4–4, and with some substrates it exceeds the wild-type activity. The successful redesign of GST A1–1 should not be generalized to suggest that supplementary mutations remote from the active site might not be necessary to reach maximal catalytic potential in other cases. Several studies indicate that rational modifications of first-sphere interactions in substrate-binding sites are insufficient (20, 40, 41), and a current approach to optimization involves addition of mutations by sequence recombinations and random mutagenesis (1, 42–45). However, in the present GST A1–1 mutant, GIMFhelix, much future improvement of the alkenal activity cannot be expected, because the activity is already approaching the highest values known with any GST substrate (6, 7). In conclusion, this investigation shows that a rational redesign of structural modules forming the substrate-binding site of an enzyme may indeed be sufficient to obtain the desired catalytic activity.
Acknowledgments
We thank Marianne Ridderström, Ina Hubatsch, Gun Stenberg, and Maryam Etahadieh from our laboratory for valuable discussions. This work was supported by grants from The Swedish Natural Science Research Council and the Swedish Research Council for Engineering Sciences.
Abbreviations
- CDNB
1-chloro-2,4-dinitrobenzene
- HHE
4-hydroxyhexenal
- HNE
4-hydroxynonenal
- GST
glutathione transferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.150084897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.150084897
References
- 1.Arnold F H, Volkov A A. Curr Opin Chem Biol. 1999;3:54–59. doi: 10.1016/s1367-5931(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 2.Sinning I, Kleywegt G J, Cowan S W, Reinemer P, Dirr H W, Huber R, Gilliland G L, Armstrong R N, Ji X, Board P G, et al. J Mol Biol. 1993;232:192–212. doi: 10.1006/jmbi.1993.1376. [DOI] [PubMed] [Google Scholar]
- 3.Mannervik B, Carlberg I, Larson K. In: Coenzymes and Cofactors. Dolphin D, Poulson R, Avramovic O, editors. 3B. New York: Wiley; 1989. pp. 475–516. [Google Scholar]
- 4.Mannervik B, Jensson H. J Biol Chem. 1982;257:9909–9912. [PubMed] [Google Scholar]
- 5.Josephy P D, Mannervik B, Ortiz de Montellano P. Molecular Toxicology. New York: Oxford Univ. Press; 1997. pp. 152–186. [Google Scholar]
- 6.Mannervik B. Adv Enzymol Relat Areas Mol Biol. 1985;57:357–417. doi: 10.1002/9780470123034.ch5. [DOI] [PubMed] [Google Scholar]
- 7.Ketterer B. Free Radical Res. 1998;28:647–658. doi: 10.3109/10715769809065820. [DOI] [PubMed] [Google Scholar]
- 8.Tsuchida S, Sato K. Crit Rev Biochem Mol Biol. 1992;27:337–384. doi: 10.3109/10409239209082566. [DOI] [PubMed] [Google Scholar]
- 9.Palinski W, Rosenfeld M E, Ylä-Herttuala S, Gurtner G C, Socher S S, Butler S W, Parthasarathy S, Carew T E, Steinberg D, Witztum J L. Proc Natl Acad Sci USA. 1989;86:1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awasthi S, Srivastava S K, Piper J T, Singhal S S, Chaubey M, Awasthi Y C. Am J Clin Nutr. 1996;64:761–766. doi: 10.1093/ajcn/64.5.761. [DOI] [PubMed] [Google Scholar]
- 11.Baez S, Segura-Aguilar J, Widersten M, Johansson A-S, Mannervik B. Biochem J. 1997;324:25–28. doi: 10.1042/bj3240025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stenberg G, Ridderström M, Engström Å, Pemble S E, Mannervik B. Biochem J. 1992;284:313–319. doi: 10.1042/bj2840313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hubatsch I, Ridderström M, Mannervik B. Biochem J. 1998;330:175–179. doi: 10.1042/bj3300175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berhane K, Widersten M, Engström Å, Kozarich J W, Mannervik B. Proc Natl Acad Sci USA. 1994;91:1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chasseaud L F. Adv Cancer Res. 1979;29:175–274. doi: 10.1016/s0065-230x(08)60848-9. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong R N. Chem Res Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 17.Bruns C M, Hubatsch I, Ridderström M, Mannervik B, Tainer J A. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 18.Scrutton N S, Berry A, Perham R N. Nature (London) 1990;343:38–43. doi: 10.1038/343038a0. [DOI] [PubMed] [Google Scholar]
- 19.Chen R, Greer A, Dean A M. Proc Natl Acad Sci USA. 1995;92:11666–11670. doi: 10.1073/pnas.92.25.11666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hedstrom L, Szilagyi L, Rutter W J. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 21.Broun P, Shanklin J, Whittle E, Somerville C. Science. 1998;282:1315–1317. doi: 10.1126/science.282.5392.1315. [DOI] [PubMed] [Google Scholar]
- 22.Jez J M, Penning T M. Biochemistry. 1998;37:9695–9703. doi: 10.1021/bi980294p. [DOI] [PubMed] [Google Scholar]
- 23.Onuffer J J, Kirsch J F. Protein Sci. 1995;4:1750–1757. doi: 10.1002/pro.5560040910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polgár L, Bender M L. J Am Chem Soc. 1966;88:3153–3154. [Google Scholar]
- 25.Neet K E, Koshland D E., Jr Proc Natl Acad Sci USA. 1966;56:1606–1611. doi: 10.1073/pnas.56.5.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Z-P, Hilvert D. J Am Chem Soc. 1990;112:5647–5648. [Google Scholar]
- 27.Stenberg G, Björnestedt R, Mannervik B. Protein Expression Purif. 1992;3:80–84. doi: 10.1016/1046-5928(92)90060-a. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson A, Mannervik B. J Mol Biol. 1999;288:787–800. doi: 10.1006/jmbi.1999.2712. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Widersten M, Björnestedt R, Mannervik B. Biochemistry. 1994;33:11717–11723. doi: 10.1021/bi00205a007. [DOI] [PubMed] [Google Scholar]
- 31.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 32.Habig W H, Pabst M J, Jakoby W B. J Biol Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 33.Mannervik B, Widersten M. In: Advances in Drug Metabolism in Man. Pacifici G M, Fracchia G N, editors. Luxembourg: European Commission; 1995. pp. 407–459. [Google Scholar]
- 34.Danielson U H, Esterbauer H, Mannervik B. Biochem J. 1987;247:707–713. doi: 10.1042/bj2470707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Björnestedt R, Stenberg G, Widersten M, Board P G, Sinning I, Jones T A, Mannervik B. J Mol Biol. 1995;247:765–773. doi: 10.1016/s0022-2836(05)80154-8. [DOI] [PubMed] [Google Scholar]
- 36.Esterbauer H, Schaur R J, Zollner H. Free Radical Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 37.Stenberg G, Board P G, Mannervik B. FEBS Lett. 1991;293:153–155. doi: 10.1016/0014-5793(91)81174-7. [DOI] [PubMed] [Google Scholar]
- 38.Björnestedt R, Tardioli S, Mannervik B. J Biol Chem. 1995;270:29705–29709. doi: 10.1074/jbc.270.50.29705. [DOI] [PubMed] [Google Scholar]
- 39.Cameron A D, Sinning I, L'Hermite G, Olin B, Board P G, Mannervik B, Jones T A. Structure (London) 1995;3:717–727. doi: 10.1016/s0969-2126(01)00206-4. [DOI] [PubMed] [Google Scholar]
- 40.Oue S, Okamoto A, Yano T, Kagamiyama H. J Biol Chem. 1999;274:2344–2349. doi: 10.1074/jbc.274.4.2344. [DOI] [PubMed] [Google Scholar]
- 41.Patten P A, Gray N S, Yang P L, Marks C B, Wedemayer G J, Boniface J J, Stevens R C, Schultz P G. Science. 1996;271:1086–1091. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- 42.Harayama S. Trends Biotechnol. 1998;16:76–82. doi: 10.1016/s0167-7799(97)01158-x. [DOI] [PubMed] [Google Scholar]
- 43.Crameri A, Dawes G, Rodriguez E, Jr, Silver S, Stemmer W P C. Nat Biotechnol. 1997;15:436–438. doi: 10.1038/nbt0597-436. [DOI] [PubMed] [Google Scholar]
- 44.Stemmer W P C. Nature (London) 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 45.Altamirano M M, Blackburn J M, Aguayo C, Fersht A R. Nature (London) 2000;403:617–622. doi: 10.1038/35001001. [DOI] [PubMed] [Google Scholar]