

Abstract
The bacterial pathogens of the genus Yersinia deliver several virulence factors into target cells using a type III secretion system. We demonstrate that Yersinia protein kinase A (YpkA), an essential bacterial virulence factor, is produced as an inactive serine/threonine kinase. The inactive kinase is activated within the host cell by a cytosolic eukaryotic activator. Using biochemical purification techniques, we demonstrate that actin is a cellular activator of YpkA. This stimulation of YpkA kinase activity by actin depends on the presence of the C-terminal twenty amino acids of YpkA, because deletion of these 20 aa not only obliterates YpkA activity, but it also destroys the interaction between YpkA and actin. Activated YpkA functions within cultured epithelial cells to disrupt the actin cytoskeleton. The disruption of the actin cytoskeleton by YpkA would be expected to inhibit macrophage function and phagocytosis of Yersinia.
Yersinia spp. (Y. pestis, Y. pseudotuberculosis, Y. enterocolitica) cause a variety of diseases including the bubonic plague. Yersinia and numerous other bacterial pathogens use a Type III secretion system to translocate effector proteins into host cells (1). These Yersinia effectors, which are referred to as Yops (Yersinia outer proteins), include YopH, YopJ, YopE, YopT, YopM, and YpkA. YopH is a protein tyrosine phosphatase responsible for inhibiting antigen presentation and disrupting focal adhesions (2–6). YopJ inhibits kinase signaling pathways and cytokine production in host cells (7–9). YopE and YopT are proteins that have effects on cellular morphology (10, 11), and YopM is a leucine-rich repeat protein with unknown function. Yersinia protein kinase A (YpkA; also known as YopO) is a protein with sequence similarity to the mammalian serine/threonine protein kinases (12) and is the only serine/threonine kinase effector identified to date. When YpkA translocates into macrophages, the cells “round up” but do not detach from the extracellular matrix (13). YpkA is essential for virulence, because disruption of the catalytic domain of YpkA results in an avirulent strain of Yersinia (12). Surprisingly, YpkA is initially produced as an inactive kinase that is activated on translocation into the host cell. We demonstrate that actin is the cellular activator of YpkA and, furthermore, that expression of YpkA in cultured epithelial cells results in a complete disruption of the actin cytoskeleton. This disruption leads to inhibition of macrophage function and phagocytosis, thereby neutralizing the innate immune response.
Materials and Methods
Constructs.
The Y. enterocolitica YpkA (YopO) ORF was isolated by PCR using the plasmid pYV8081 (a generous gift from James Bliska, State University of New York at Stony Brook). The oligonucleotides used were as follows: 5′-ACTGGAATTCTATCACCTTCGATCTCCCTCG-3′ (sense) and 5′-CATCAGCTCGAGTCACATCCATTCCCGCTC-3′ (antisense). These oligonucleotides allowed for insertion into the EcoRI/XhoI restriction sites and in-frame fusion with glutathione S-transferase (GST) in the vector pGEX-KG (14). YpkA was subcloned by using the EcoRI/XhoI fragment into the Prescission Protease System Vector pGEX-6p-2 (Amersham Pharmacia), allowing for in-frame fusion with GST and insertion of a Prescission protease cleavage site for the production of soluble YpkA. YpkA was also subcloned into the mammalian expression vector pcDNA3 (Invitrogen) for transfection into human epithelial kidney (HEK) 293 cells under the control of the human cytomegalovirus (CMV) promoter. Full-length YpkA and several C-terminal deletions were amplified by using PCR and the following oligonucleotide pairs: full-length N-terminal FLAG tag YpkA, 5′-GATCGAATTCACCATGGACTACAAGGACGACGATGACAAGTCACCTTCGATCTCCCTCG-3′ (sense)/5′-TCAGTCTAGATCACATCCATTCCCGCTC-3′ (antisense); Δ710–730 N-terminal FLAG tag YpkA, sense oligonucleotide same as full-length N-terminal FLAG tag YpkA/5′-GCTAATTCTAGATCATCTCTGTTCAACCAAATGCT-3′ (antisense); Δ660–730 N-terminal FLAG tag YpkA, sense oligonucleotide same as full-length N-terminal FLAG tag YpkA/5′-GCTAATTCTAGATCAGTGAATTGCGGTATACTGC-3′ (antisense); Δ430–730 N-terminal FLAG tag YpkA, sense oligonucleotide same as full-length N-terminal FLAG tag YpkA/5′-GCTAATTCTAGATCACAAATCAGAGAGAGCTCCCG-3′ (antisense); full-length C-terminal FLAG tag YpkA, 5′-CTAGCTAGCTCGAGTCCCATCCATTCCCGCTCCAAC-3′ (sense)/5′-CATGCATGGGTACCGCCGCCATGTCACCTTCGATCTCCCTCG-3′ (antisense). The N-terminal FLAG tag YpkA (full-length and C-terminal deletions) was inserted into the EcoRI/XbaI restriction sites of pcDNA3, and the C-terminal FLAG tag YpkA was inserted into the XhoI/KpnI restriction sites of pcDNA3. The mutation K269A in all vectors was made by using the Quik-Change Site-directed mutagenesis protocol (Stratagene). The oligonucleotide pair used was 5′-CATAACGATATCGCACCCGGGAATGTGGTA-3′ (sense)/5′-TACCACATTCCCGGGTGCGATATCGTTATG-3′ (antisense). All constructs were sequenced for verification of wild-type or mutant sequence.
Expression of Recombinant YpkA.
The vector pGEX-6p-2-YpkA was transformed into Novablue DE3 Escherichia coli-competent cells (Stratagene) as previously described (15). Cells were inoculated into 1 liter of 2 x YT containing 100 μg/ml ampicillin and allowed to grow to a density of 0.5 as measured by OD600. Cells were then shifted to room temperature, and protein expression was induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. Cells were harvested and resuspended in lysis buffer (30 mM Tris, pH 7.4/150 mM NaCl/10% (vol/vol) glycerol/0.1% (vol/vol) Triton X-100/0.1 mM PMSF). Cells were lysed by two passages through a French press, and bacterial debris was pelleted at 20,000 × g for 20 min. The supernatant was then applied to 5 ml of glutathione agarose (50% slurry in lysis buffer). GST-YpkA glutathione agarose was washed six times with lysis buffer and stored at 4°C. YpkA was eluted off of the glutathione agarose by using 3 ml of lysis buffer containing 200 units Prescission protease (Amersham Pharmacia) and incubating overnight at 4°C. Concentrations of YpkA were determined by Coomassie blue staining of SDS/PAGE gels using BSA standards.
Kinase Assay.
YpkA autophosphorylation or phosphorylation of artificial substrates was measured by using an in vitro kinase assay. Recombinantly expressed YpkA (1.5 μg) and HeLa cell extract [3 μg of protein as determined by Bradford assay (Bio-Rad)] were added in an in vitro kinase assay containing 20 mM Hepes (pH 7.4), 10 mM magnesium acetate, 1 mM DTT, 0.5 mM ATP, and 2 μCi [γ-32P]ATP (New England Nuclear). The reaction was incubated at 30°C for 30 min and quenched with loading buffer (250 mM Tris, pH 6.8/40% (vol/vol) glycerol/20% (vol/vol) 2-mercaptoethanol/8% (wt/vol) SDS/0.01% Bromophenol blue). One-third of the kinase reaction was electrophoresed on a 12% SDS/PAGE gel and transferred to Immobilon-P nitrocellulose (Millipore) for 1 h at 100 V. Radioactivity was analyzed by autoradiography. YpkA was expressed in HEK293 cells by transient transfection of the appropriate pcDNA3–YpkA construct by using FuGENE 6 (Roche Molecular Biochemicals). Cells were lysed in TNN buffer (50 mM Tris, pH 8.0/150 mM NaCl/1% (vol/vol) Nonidet P-40). YpkA was immunoprecipitated from lysates by using an anti-FLAG M2 affinity gel (Sigma). The affinity gel was washed three times with TNN buffer, and immunoprecipitates were assayed by in vitro kinase assay as described previously (see above). Protein levels of YpkA in immunoprecipitates were analyzed by Western blot using an anti-FLAG antibody. The concentration of artificial substrates used in the in vitro kinase assays with YpkA are as follows: 50 μg/ml myelin basic protein; 50 μg/ml Elk; 0.5 mg/ml phosvitin; 0.1 mg/ml histone; 0.5 mg/ml casein; 50 μg/ml Phas 1; and 50 μg/ml Fos.
Preparation of Mammalian Extracts.
HeLa cells were cultured in DMEM containing 10% (vol/vol) FCS, 2 mM glutamine, and 100 μg/ml penicillin/streptomycin (GIBCO). Approximately 107 cells were washed once with PBS and resuspended in 25 ml of homogenization buffer (10 mM Tris, pH 7.5/10% (wt/vol) sucrose/2 μg/ml aprotinin/2 μg/ml leupeptin/0.2 mM PMSF). Cells were homogenized with 30 passes in a Dounce homogenizer. The cell lysate was pelleted twice at 5,000 × g for 20 min to collect unbroken nuclei. The supernatant was spun at 100,000 × g for 30 min to collect the cytosolic (supernatant) and membrane (pellet) fractions. The membrane fraction was resuspended in homogenization buffer and sonicated for solubilization.
Bovine brain cytosolic proteins were isolated as follows. Bovine brains were homogenized in 1 volume TEDN buffer (20 mM Tris, pH 7.4/100 mM NaCl/1 mM EDTA/1 mM DTT/1 μg/ml aprotinin/1 μg/ml leupeptin/0.1 mM PMSF). Soluble proteins were isolated through centrifugation at 100,000 × g.
Purification of a YpkA Activator.
Soluble bovine brain proteins were separated over a Q-Sepharose anion exchange column using TEDN buffer with a salt gradient from 100 mM NaCl to 500 mM NaCl. Fractions were assayed for their ability to activate recombinant YpkA by autophosphorylation in an in vitro kinase assay (see above). These fractions were pooled, brought to a concentration of 1 M ammonium sulfate, and separated over a Phenyl Sepharose hydrophobic interaction column using TEDN buffer with an ammonium sulfate gradient from 1 M ammonium sulfate to 0 M ammonium sulfate. Positive fractions were pooled and separated over a Sephacryl 200 High Resolution gel filtration column (3.8 cm × 85.1 cm) using TEDN buffer. Proteins from this final purification step contained in the positive fractions were separated by SDS/PAGE and analyzed by using silver stain (16).
Cell Culture.
HEK293 and HeLa cells were transfected by using the FuGENE 6 transfection kit (Roche Molecular Biochemicals). After 24 h, cells were either lysed with TNN (50 mM Tris⋅HCl, pH 8.0/150 mM NaCl/1% Nonidet P-40) containing protease inhibitors (Complete; Roche Molecular Biochemicals) or stained for actin filaments.
Preparation of DNase I-Sepharose.
DNase I (Worthington) was conjugated to cyanogen bromide-activated Sepharose (Amersham Pharmacia) following manufacturer's instructions. Briefly, 2.5 mg of DNase I was dissolved in coupling buffer (0.1 M NaHCO3, pH 8.3/0.5 M NaCl). Cyanogen bromide-activated Sepharose was washed with 1 mM HCl, and DNase I was added and incubated overnight at 4°C. The beads were washed with coupling buffer and incubated in 0.1 M Tris⋅HCl (pH 8.0) for 2 h at 4°C. Sepharose was washed with three cycles of alternating pH buffers A and B (buffer A, 0.1 M sodium acetate, pH 4.0/0.5 M NaCl; buffer B, 0.1 M Tris⋅HCl, pH 8.0/0.5 M NaCl).
Cell Microscopy.
HeLa cells expressing YpkA were washed with PBS and fixed in 4% paraformaldehyde in PBS for 10 min. Cells were washed with PBS and permeabilized with 0.2% Triton X-100 in PBS. Cells were washed with PBS, and actin filaments were stained for 25 min by using 33 μM rhodamine phalloidin (Molecular Probes) in PBS. Cells were mounted on slides with Crystal Mount (Biomeda).
Results and Discussion
Identification of Actin as the Eukaryotic Activator of YpkA.
To develop a better understanding of the physiological function of YpkA, we expressed and purified the recombinant kinase from E. coli. Galyov et al. have demonstrated that YpkA secreted by Y. pseudotuberculosis possesses a low level of serine/threonine kinase activity; however, our recombinant YpkA was unable to undergo autophosphorylation (Fig. 1A, lane 1). In contrast, when the recombinant protein was added to a HeLa cell extract, the kinase was capable of autophosphorylation (Fig. 1A, lane 2). This observation suggested that the eukaryotic cell extract might contain an activator of the kinase. To further characterize the eukaryotic activator, YpkA was expressed and immunoprecipitated from human embryonic kidney (HEK293) cells. The immunoprecipitated protein displayed a high level of kinase activity (Fig. 1B, lane 1). YpkA produced from HEK293 cells was also capable of phosphorylating other protein substrates, such as histones and myelin basic protein (Fig. 1C). YpkA seems to preferentially phosphorylate basic substrates, such as histones and myelin basic protein, in contrast to neutral and acidic substrates, such as Elk, phosvitin, casein, Phas 1, and Fos. This observation suggests that YpkA is likely to recognize serine and/or threonine residues within the context of a basic region within the protein. To determine whether the observed kinase activity was associated with YpkA and was not a trans-phosphorylation event involving a kinase present in the HEK293 cells, a lysine residue in the catalytic loop of YpkA was mutated to alanine (YpkA-K269A). Transient expression of the protein encoded by this plasmid in HEK293 cells followed by immunoprecipitation demonstrated that YpkA-K269A was attenuated in its ability to autophosphorylate as well as phosphorylate myelin basic protein (Fig. 1B, lanes 3 and 4). Collectively, these observations suggest that HEK293 cells, as well as HeLa cell extracts, contain an activator of YpkA.
Figure 1.
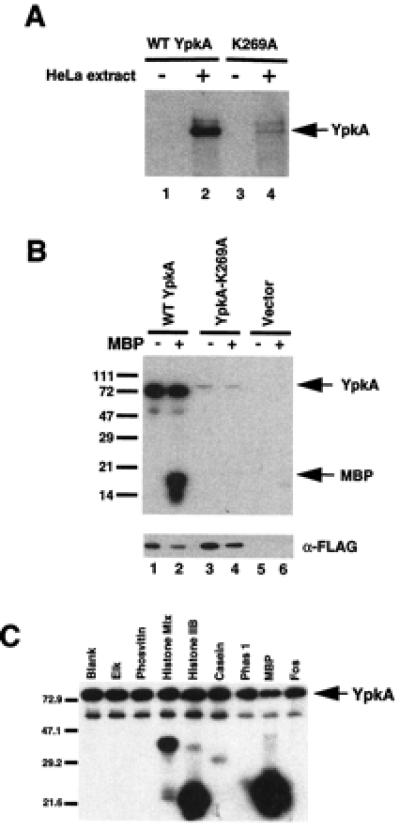
YpkA requires a protein cofactor for activation. (A) Recombinant YpkA requires an activator for kinase activity. Recombinant YpkA or YpkA-K269A was expressed in E. coli and purified. YpkA was incubated with either buffer or HeLa cell extract protein (3 μg) and subjected to in vitro kinase assays. (B) HEK293 cells were transfected with either wild-type YpkA, YpkA-K269A, or vector (8 μg). Both wild-type and mutant YpkA were N-terminal FLAG epitope tagged. YpkA was immunoprecipitated with anti-FLAG M2 affinity gel (Sigma) and was subjected to in vitro kinase assays. Similar levels of YpkA are present in all of the immunoprecipitation extracts as determined by immunoblot analysis with an antibody to the FLAG epitope (Sigma). (C) HEK293 cells were transfected with wild-type YpkA (8 μg). YpkA was immunoprecipitated with anti-FLAG M2 affinity gel (Sigma) and was subjected to in vitro kinase assays in the presence of the artificial substrates shown, as described in Materials and Methods.
To identify the activator of YpkA, we prepared extracts from two cell lines (HEK293, HeLa), as well as from rat tissues (spleen, liver, kidney, brain), bovine brain, and yeast cells, and added these extracts to our inactive recombinant kinase. Similar results to those shown in Fig. 1A for the soluble extract from HeLa cells, which result in YpkA activation, were obtained by using soluble extracts from other cells and tissues (data not shown). Likewise, the addition of soluble extract to recombinant mutant YpkA-K269A had little effect on kinase activation (Fig. 1A, lanes 3 and 4). Boiling or trypsin treatment of the soluble extract rendered it incapable of stimulating YpkA activity, suggesting that the activator is proteinaceous in nature (data not shown).
We initiated our purification of the YpkA-stimulating activity from bovine brain cytosol. Proteins were sequentially fractionated by using Q Sepharose Fast Flow anion exchange chromatography, Phenyl Sepharose hydrophobic interaction chromatography, and Sephacryl 200 High Resolution gel filtration chromatography. Fractions were assayed for their ability to activate the bacterially expressed YpkA. Fractions possessing YpkA stimulatory activity from the final step in the purification were then analyzed by SDS/PAGE (Fig. 2A). Several proteins were identified as candidate activators of YpkA activity, including a 12-kDa and a 45-kDa protein (Fig. 2A). The 12-kDa protein has been identified as profilin by mass spectrometry (C. Moomaw and C. Slaughter, unpublished data); however, purified profilin was incapable of activating YpkA (data not shown). The 45-kDa protein had an apparent molecular weight similar to that of actin, which also had an elution profile from the Sephacryl 200 gel filtration column similar to that of the activator (Fig. 2A). Based on these observations, we tested purified bovine actin for its ability to activate recombinant YpkA. Purified bovine actin stimulated recombinant YpkA in a dose-dependent fashion (Fig. 2B). This dose-dependent YpkA activation required intrinsic YpkA kinase activity because YpkA-K269A was inactive under comparable conditions (data not shown). This observation excludes the possibility of a trans-phosphorylation event catalyzed by a contaminating exogenous kinase present in the purified actin. Although G-actin was the major form purified from bovine brain cytosol, preliminary data suggest that both F- and G-actin are capable of activating YpkA (data not shown). Furthermore, depletion of actin from the partially purified Sephacryl 200 gel filtration column fraction using an actin binding resin, DNase I-Sepharose (17, 18), resulted in a stepwise decrease in the YpkA activation potential by the resulting partially depleted fraction (Fig. 2C). As expected, the DNase I-Sepharose-bound actin was also capable of activating YpkA (Fig. 2C). Taken together, these results demonstrate that actin is a cellular activator of YpkA. To our knowledge, there has not been any other evidence of a bacterial protein kinase activated by actin within the host cell. Fig. 2A also suggests that, in addition to actin serving as an activator of YpkA, actin can also function as an in vitro substrate of the kinase.
Figure 2.
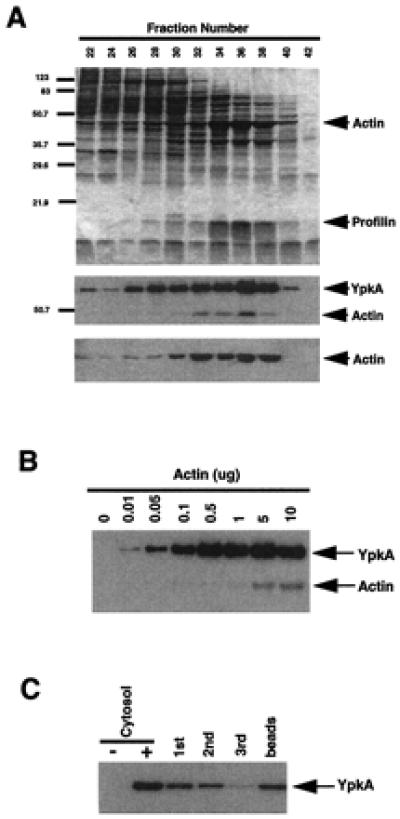
The activator of YpkA kinase activity is actin. (A) Partial purification of the YpkA activator. Bovine brain cytosol proteins were separated by column chromatography. Proteins contained within the peak fractions collected from the Sephacryl 200 gel filtration column were analyzed by SDS/PAGE and detected by silver staining. Eluted actin was detected by immunoblot analysis using an antibody to actin (Sigma). (B) Dose-dependent activation of YpkA by actin. Actin was added in increasing concentrations to recombinant YpkA (1.5 μg). Actin is also phosphorylated by YpkA in vitro. (C) Actin was depleted from the Sephacryl 200 gel filtration peak fraction by using successive incubations with DNase I-Sepharose as previously described. Fractions were assayed for their ability to stimulate YpkA kinase activity.
The C Terminus of YpkA Is Essential for the Interaction Between YpkA and Actin.
Previously, it has been observed that Yersinia lacking the C terminus of YpkA are avirulent (12). To understand why the C terminus is essential for virulence, we prepared several C-terminal deletions of a FLAG-tagged YpkA. The largest of these C-terminal deletions leaves the entire kinase domain intact (residues 1–450) (Fig. 3A). Each of these constructs was expressed in HEK293 cells, and kinase assays were performed with immunoprecipitated YpkA. Deletion of only 20 aa from the C terminus (Δ710) resulted in complete loss of kinase activity (Fig. 3B). As expected, subsequent deletions of the kinase C terminus (Δ450 or Δ660) could not be activated (Fig. 3B). Furthermore, only full-length YpkA and YpkA-K269A were capable of interacting with actin as assessed by coimmunoprecipitation (Fig. 3B). Because there are no serine residues within the C-terminal 20 aa of YpkA, the effects of this truncation (YpkA-Δ710) on autophosphorylation are not due to removal of a critical phosphorylation site (12). These results demonstrate that actin binding is mediated principally by the C-terminal 20 aa of YpkA, and suggest that actin binding leads to YpkA activation.
Figure 3.
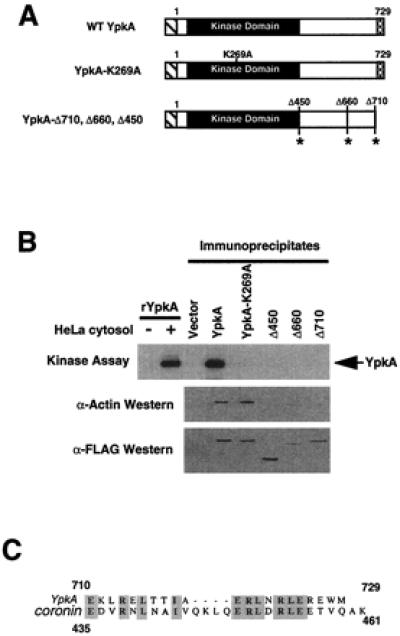
YpkA requires the last 20 aa for activation. (A) Schematic representation of YpkA constructs. Slashed lines denote the FLAG epitope tag. The C-terminal 20 aa are shown in black and white checkered boxes. Each of the deletion mutants is labeled, and the site of the C-terminal truncation is denoted with an asterisk. (B) YpkA requires the last 20 aa for kinase activity. HEK293 cells were transfected with YpkA, YpkA-K269A, YpkA-Δ450, YpkA-Δ660, YpkA-Δ710, or pCDNA3 (8 μg). All YpkA constructs were fused to an N-terminal FLAG epitope tag. YpkA was immunoprecipitated with anti-FLAG M2 affinity gel (Sigma) and subjected to in vitro kinase assays. Actin was detected in immunoprecipitates using an antibody against actin (Sigma). Levels of YpkA protein expression were assessed by immunoblot analysis with an antibody to the FLAG epitope (Sigma). (C) The C termini of YpkA and coronin are similar. Sequence alignment was performed by using the clustalw algorithm in the macvector program. Identical residues are highlighted in dark gray, whereas similar residues are highlighted in light gray.
YpkA Disrupts the Actin Cytoskeleton in Vivo.
During infection, YpkA has been shown to cause cells to “round up” while maintaining contact at focal adhesions (13). To understand the effect that YpkA has on actin dynamics in vivo, HeLa cells were cotransfected with a plasmid encoding the enhanced green fluorescent protein and either vector alone or an expression vector encoding wild-type YpkA, YpkA-K269A, or YpkA-Δ710. These cells were subsequently fixed and stained with rhodamine phalloidin to visualize the microfilament system. Cells expressing YpkA had a pronounced phenotype (Fig. 4C), and, although these cells appear to be sending out extensions, examination over time of these changes indicates that they are actually retracting their edges (i.e., “rounding up”) while maintaining focal adhesion contacts, similar to the morphology observed in cells infected with Yersinia overexpressing YpkA (13). Strikingly, cells expressing wild-type YpkA did not have detectable actin stress fibers (Fig. 4D), indicating that the kinase has completely disrupted the microfilament network in these cells. Expression of the kinase mutant, YpkA-K269A, resulted in an intermediate phenotype (Fig. 4, E and F). These cells had an abnormal morphology, demonstrated by the presence of small retraction fibers, but the actin microfilament system was not completely obliterated, as seen in cells expressing wild-type YpkA. The morphology of cells in which the YpkA-K269A mutant was expressed may be the result of its very low-level kinase activity. Alternatively, effects of the YpkA-K269A mutant may be mediated by the C terminus of the inactive kinase competing for a binding site with proteins playing a role in actin bundling or polymerization. To explore this possibility, we compared the sequence of the C-terminal 20 aa of YpkA with all ORFs present in the nonredundant data bank by using Ψ-blast. The C terminus of YpkA has significant sequence identity to the C terminus of coronin, a known actin-binding protein (Fig. 3C). The C terminus of coronin is necessary for the bundling of actin fibers (19). In addition, the C terminus of coronin forms an amphipathic helix similar to the predicted secondary structure of the last 20 aa of YpkA. The similarity between the C-terminal domains of YpkA and coronin raises the intriguing possibility that the YpkA-K269A mutant contributes to the disruption of actin filaments by direct competition with coronin for the same binding site on actin. The hypothesis that YpkA may occupy the coronin-binding site on actin is also supported by the observation that cells expressing YpkA-Δ710 were indistinguishable from controls in their morphology and cytoskeleton (Fig. 4, G and H). As expected, cells that are not expressing YpkA displayed a normal array of stress fibers, suggesting that the microfilament structures within these cells are intact (Fig. 4, A and B). Our results demonstrate that YpkA has an in vivo global effect on the actin cytoskeleton and that this effect requires both kinase activity and the presence of an intact YpkA C terminus. Because actin is crucial in the innate immune system for its role in both phagocytosis and chemotaxis, the disruption of the actin cytoskeleton by YpkA would be predicted to inhibit these processes, thus ensuring survival of Yersinia within the host.
Figure 4.
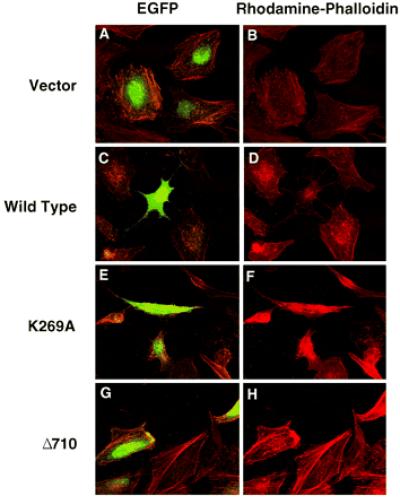
YpkA overexpression causes actin filament disruption. HeLa cells were cotransfected with a vector encoding the enhanced green fluorescent protein (250 ng) and either YpkA, YpkA-K269A, YpkA-Δ450, YpkA-Δ660, YpkA-Δ710, or pCDNA3 (1.75 μg). Cell morphology was visualized by expression of enhanced green fluorescent protein. Actin filaments were visualized by staining with rhodamine phalloidin. Pictures shown are representative of all transfected cells stained.
We have demonstrated that the Yersinia protein kinase is produced in bacteria as an inactive catalyst. On translocation into the mammalian cell by the type III secretory system, YpkA localizes to the inner surface of the plasma membrane (13). This localization would place YpkA near the actin cortex, where, on activation, the kinase could phosphorylate a number of proteins that play key roles in regulating the actin cytoskeleton. This phosphorylation in turn results in a catastrophic depolymerization of cellular actin filaments that has a profound effect on macrophages and other cells functioning in the innate immune system. Recently, much has been learned about the actin cytoskeleton as a result of the identification of another bacterial virulence factor from Listeria, ActA. ActA is required by Listeria for mobility within the host cell and between host cells. ActA functions to assemble filamentous actin on its surface within the cytoplasm of the target cell (20, 21). We anticipate that the identification of the YpkA substrates may also lead to a better understanding of the processes governing actin dynamics within the cell.
Acknowledgments
We thank Matt Wishart, Gregory Taylor, Tomohiko Maehama, Vic DiRita, and Susan Brown for their thoughts and insights on this manuscript. We also thank Zhao Qin Bao, Michael Roh, Carolyn Moomaw, and Clive Slaughter for their generous technical assistance. We also thank Ilana Nodulman and Clarence Schutt (Princeton University) for their generous gift of purified profilins. This work was funded by National Institutes of Health (NIH) Grant 18849 (to J.E.D.), NIH Molecular Mechanisms of Microbial Pathogenesis Training Grant AI07528 (to S.J.J.), and the Walther Cancer Institute (to J.E.D.).
Abbreviations
- YpkA
Yersinia protein kinase A
- Yop
Yersinia outer protein
- HEK
human embryonic kidney
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170281997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170281997
References
- 1.Cornelis G R, Boland A, Boyd A P, Geuijen C, Iriarte M, Neyt C, Sory M P, Stainier I. Microbiol Mol Biol Rev. 1998;62:1315–1352. doi: 10.1128/mmbr.62.4.1315-1352.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan K L, Dixon J E. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- 3.Bliska J B, Copass M C, Falkow S. Infect Immun. 1993;61:3914–3921. doi: 10.1128/iai.61.9.3914-3921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black D S, Bliska J B. EMBO J. 1997;16:2730–2744. doi: 10.1093/emboj/16.10.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosqvist R, Bolin I, Wolf-Watz H. Infect Immun. 1988;56:2139–2143. doi: 10.1128/iai.56.8.2139-2143.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persson C, Carballeira N, Wolf-Watz H, Fallman M. EMBO J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orth K, Palmer L E, Bao Z Q, Stewart S, Rudolph A E, Bliska J B, Dixon J E. Science. 1999;285:1920–1923. doi: 10.1126/science.285.5435.1920. [DOI] [PubMed] [Google Scholar]
- 8.Mills S D, Boland A, Sory M P, van der Smissen P, Kerbourch C, Finlay B B, Cornelis G R. Proc Natl Acad Sci USA. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monack D M, Mecsas J, Ghori N, Falkow S. Proc Natl Acad Sci USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosqvist R, Forsberg A, Wolf-Watz H. Infect Immun. 1991;59:4562–4569. doi: 10.1128/iai.59.12.4562-4569.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iriarte M, Cornelis G R. Mol Microbiol. 1998;29:915–929. doi: 10.1046/j.1365-2958.1998.00992.x. [DOI] [PubMed] [Google Scholar]
- 12.Galyov E E, Hakansson S, Forsberg A, Wolf-Watz H. Nature (London) 1993;361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- 13.Hakansson S, Galyov E E, Rosqvist R, Wolf-Watz H. Mol Microbiol. 1996;20:593–603. doi: 10.1046/j.1365-2958.1996.5251051.x. [DOI] [PubMed] [Google Scholar]
- 14.Guan K L, Dixon J E. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 16.Blum H, Beier H, Gross H J. Electrophoresis. 1987;8:93–99. [Google Scholar]
- 17.Podolski J L, Steck T L. J Biol Chem. 1988;263:638–645. [PubMed] [Google Scholar]
- 18.Lazarides E, Lindberg U. Proc Natl Acad Sci USA. 1974;71:4742–4746. doi: 10.1073/pnas.71.12.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cossart P, Kocks C. Mol Microbiol. 1994;13:395–402. doi: 10.1111/j.1365-2958.1994.tb00434.x. [DOI] [PubMed] [Google Scholar]
- 20.Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wachter M, Wuenscher M, Chakraborty T. EMBO J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]