

Abstract
The developmentally important homeodomain transcription factors of the NK-2 class contain a highly conserved region, the NK2-specific domain (NK2-SD). The function of this domain, however, remains unknown. The primary structure of the NK2-SD suggests that it might function as an accessory DNA-binding domain or as a protein–protein interaction interface. To assess the possibility that the NK2-SD may contribute to DNA-binding specificity, we used a PCR-based approach to identify a consensus DNA-binding sequences for Nkx2.2, an NK-2 family member involved in pancreas and central nervous system development. The consensus sequence (TCTAAGTGAGCTT) is similar to the known binding sequences for other NK-2 homeodomain proteins, but we show that the NK2-SD does not contribute significantly to specific DNA binding to this sequence. To determine whether the NK2-SD contributes to transactivation, we used GAL4-Nkx2.2 fusion constructs to map a powerful transcriptional activation domain in the C-terminal region beyond the conserved NK2-SD. Interestingly, this C-terminal region functions as a transcriptional activator only in the absence of an intact NK2-SD. The NK2-SD also can mask transactivation from the paired homeodomain transcription factor Pax6, but it has no effect on transcription by itself. These results demonstrate that the NK2-SD functions as an intramolecular regulator of the C-terminal activation domain in Nkx2.2 and support a model in which interactions through the NK2-SD regulate the ability of NK-2-class proteins to activate specific genes during development.
Developmental processes are frequently associated with the expression of specific homeodomain transcription factors. Among them, transcription factors of the NK-2 class make up a unique family. These factors are structurally related to the Drosophila NK-2 family of transcription factors (NK2/vnd, NK3/bagpipe, and NK4/tinman), which are known to play important roles during development (1). The vertebrate NK-2 factors also regulate development: Nkx2.1 is involved in the regulation of thyroid, lung, and ventral forebrain development (2, 3), and Nkx2.5 is required for proper heart formation (4, 5).
The transcription factors of this class contain three highly conserved regions: the homeodomain, the TN (tin) domain (or NK decapeptide), and the NK2-specific domain (NK2-SD; also called the NK2 domain) (1). The homeodomain is a highly conserved DNA-binding domain; the homeodomains of the NK-2 class are believed to recognize the DNA sequence 5′-CAAG-3′, whereas most other homeodomain transcription factors recognize 5′-TAAT-3′ (6, 7). The short TN domain is found at the N-terminal region of most NK-2 proteins. The NK2-SD is unique to NK-2-class proteins and is separated from the C-terminal end of the homeodomain by a short linker. The NK2-SD is composed of a hydrophobic core sequence VAVPVLV, with a central proline that prevents helix formation, and is flanked by basic amino acids. This structure suggests that the NK2-SD might function as an accessory DNA-binding domain or as a protein–protein interaction interface (8). Previous studies have mapped the functional domains of Nkx2.1 and Nkx2.5 and have found both activation and repression domains in both proteins. However, these studies did not implicate the TN domain and NK2-SD directly in the function of these proteins (9, 10).
In this study, we used Nkx2.2 as a model protein for investigating the functional role of the NK2-SD. Nkx2.2 was originally identified as a member of the NK-2 class of transcription factors expressed in the developing central nervous system (11) and pancreas (12). The targeted disruption of Nkx2.2 alters the development of the spinal cord (13) and pancreatic islet cells (14). However, the direct target genes and the specific function of Nkx2.2 are unknown. We determined the DNA-binding characteristics and transactivation properties of Nkx2.2 as they relate to the NK2-SD. Our results reveal that Nkx2.2 recognizes the DNA sequence TCTAAGTGAGCTT and that the NK2-SD does not contribute significantly to the specificity of DNA binding. The NK2-SD does contribute significantly, however, to transactivation function, inasmuch as mutation or deletion of this domain unmasks a powerful transcriptional activation function in Nkx2.2. Our results suggest a model in which Nkx2.2, regulated by interactions through the NK2-SD, causes activation of specific genes during development.
Experimental Procedures
Binding Site Selection.
The PCR-based, random oligonucleotide selection procedure was described previously (9, 15, 16). Briefly, a 32P-labeled 55-mer oligonucleotide with a central random stretch of 15 bp (5′-AGACGGATCCTATGCGCGATNNNNNNNNNNNNNNNCAGTAGCTATCTGCAGGCGT-3′) was used as a probe in binding reactions with bacterially produced 6XHis-(112–222)Nkx2.2 protein encoding the homeodomain and NK2-SD of human Nkx2.2 (17). Specifically bound 55-mers were isolated by electrophoretic mobility-shift assay (EMSA) on a polyacrylamide gel, amplified by PCR with the primers FWD (5′-AGACGGATCCTATGCGCGA-3′) and REV (5′-ACGCCTGCAGATAGCTACTG-3′), and used again in additional rounds of selection. After a total of eight rounds of selection, the resulting 55-mers were subcloned into pCR2.1 (CLONTECH) and sequenced. Sequences were analyzed using a nongapped algorithm (clustal w) in macvector 6.5 software (Oxford Molecular, Hunt Valley, MD).
EMSAs and Binding Competition.
EMSA buffers and electrophoresis conditions were as previously described (18). Nkx2.2 and its truncated derivatives were expressed in vitro from coding fragments subcloned into pBAT11 (19), using the T7 TNT Quick Coupled Reticulocyte Lysate System (Promega). One microliter of the in vitro reaction mixture was added to each binding reaction. Where nuclear extracts were used, 2 μg of the protein was used. Binding competition assays were performed under identical EMSA conditions, except that only the Nkx2.2 consensus sequence was labeled with 32P, and various concentrations (from 10−11 to 10−6 M) of unlabeled competitor sequence were added. Radioactivity in the EMSA gels was quantitated by PhosphorImager analysis (Molecular Dynamics), and the data were modeled using simple one-step binding kinetics as described previously (15).
Plasmid Construction and Site-Directed Mutagenesis.
The one-hybrid expression vectors encoding Nkx2.2-GAL4DBD fusion proteins (GAL4DBD is yeast GAL4 DNA-binding domain) were made by PCR, using human Nkx2.2 cDNA as a template, then ligating the fragments in-frame into the simian virus 40 promoter-driven GAL4DBD expression vector pM (CLONTECH). The expression vectors encoding various fragments of the Nkx2.2 cDNA driven by the cytomegalovirus (CMV) promoter were made by subcloning the Nkx2.2 cDNA fragments from the cognate pM vectors into pBAT12 (19). Mutagenesis of the Nkx2.2 coding sequence was performed with a Quick Change mutagenesis kit (Stratagene). All constructs were confirmed by sequencing.
Cell Culture and Transient Transfections.
NIH 3T3 cells, Cos7 cells, and βTC3 cells were cultured as previously described (18). For one-hybrid experiments, GAL4DBD-Nkx2.2 fusion protein expression, pFOXLuc1-G5E1b or pFOXLuc2G5prl, and CMVβgal plasmids were cotransfected into each cell with Superfect (Qiagen, Chatsworth, CA). Forty-eight hours after the transfection, cells were harvested, and luciferase and β-galactosidase assays were performed as described previously (19).
For transfection experiments using pBAT12 constructs, cells were transfected with pFOXLuc1–7xNk2prl and Nkx2.2 expression vectors, harvested 48 h later, and assayed for luciferase activity. All experiments were repeated at least three times.
Western Blot Analysis.
Expression of the GAL4-Nkx2.2 fusion protein derivatives in nuclear extracts of Cos7 cells or NIH 3T3 cells were assessed by Western blot analysis with polyclonal anti-GAL4DBD antibody (Santa Cruz Biotechnology). Western blots were visualized with the ECL-Plus system (Amersham Pharmacia Biotech).
Results
Selection of an Optimal DNA-Binding Site for Nkx2.2.
Previous studies identifying the optimal binding sites of NK-2 homeodomain proteins have used protein constructs containing the homeodomain alone (9), and none have included the NK2-SD. Indeed, the structure of the NK2-SD suggests a single loop reminiscent of the nonhelical loops in known DNA-binding domains (8). To test the possibility that NK2-SD plays a role in DNA binding, we expressed in Escherichia coli a protein containing the homeodomain and NK2-SD of Nkx2.2 (amino acids 112–222) and used the purified protein to select for binding of specific DNA sequences from among a library of oligonucleotides. The oligonucleotides, which were end-labeled with 32P, were 55 bp in length and contained a central random stretch of 15 bp. The protein and its bound DNA sequences were separated from unbound oligonucleotide by EMSA. The protein–oligonucleotide complex was cut and eluted from the polyacrylamide gel, and the oligonucleotide sequence was amplified by PCR with incorporation of radiolabel. This process was repeated a total of eight times, after which the sequences of 23 oligonucleotides were determined. The sequences were subjected to an ungapped alignment, using the multiple-sequence alignment function in the macvector 6.5 software package (Oxford Molecular). Fig. 1A demonstrates that the oligonucleotide sequences are nonrandomly distributed and contain the consensus sequence TCTAAGTGAGCTT. Fig. 1A also shows that this consensus is flanked on its 5′ and 3′ ends by a predominance of A+T-rich sequences. Fig. 1B demonstrates that an oligonucleotide containing the consensus sequence binds ()Nkx2.2, native Nkx2.2, and the homeodomain of Nkx2.2 alone (amino acids 128–187) in an EMSA.
Figure 1.
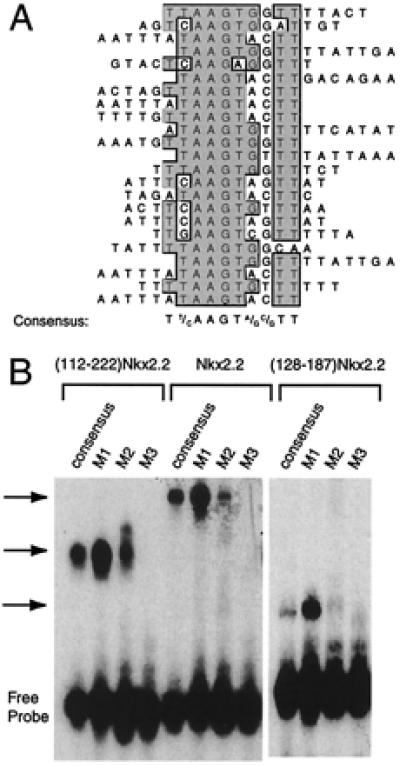
Nkx2.2 binding site selection. (A) Binding site selection was performed with the human Nkx2.2 homeodomain and NK2-SD (amino acids 112–221) and a set of oligonucleotides containing a 15-bp random stretch flanked by PCR primer sites. Sequenced products from the eighth round of selection are shown aligned by the clustal w alignment algorithm in macvector 6.5 software (Oxford Molecular). The consensus sequence that emerges from the best-fit line-up is shown. (B) An EMSA using in vitro translated ()Nkx2.2, full-length Nkx2.2, and the Nkx2.2 homeodomain alone (amino acids 128–187). Different 32P-labeled oligonucleotides (sequences shown in Table 1) were incubated with 1 μl of each in vitro translated protein for 15 min at room temperature and then subjected to electrophoresis on a 5% polyacrylamide gel. The free probe and retarded complex are indicated.
DNA-Binding Characteristics of Nkx2.2.
The consensus sequence we have determined here contains the “core” motif CTAAGTGA, which is similar to the core sequence TNNAGTG or CAAGTG determined for other Nk2 family members (7, 9). To determine if sequences within and flanking this core have significant effects on the DNA-binding affinity of ()Nkx2.2, native Nkx2.2, and the Nkx2.2 homeodomain, we performed EMSAs using these proteins and 32P-labeled oligonucleotides that contain alterations to the consensus sequence. The data in Fig. 1B show that the three proteins display parallel affinities for DNA. Importantly, the homeodomain of Nkx2.2 alone (amino acids 127–188) also displays parallel binding affinities for the oligonucleotides, demonstrating that the NK2-SD does not contribute significantly to either the specificity or the affinity of DNA interactions. A single T → C base pair mutation (oligonucleotide M1, TAAGTG → CAAGTG) is well tolerated within the core sequence, which is a predictable finding, inasmuch as both sequences were obtained in the binding site selection experiment and the consensus sites for other NK-2 family members often have a C at this position. On the other hand, other changes within the core, and flanking the core, are significantly less well tolerated (oligonucleotides M2 and M3).
To obtain quantitative information on the importance of specific base pairs within the Nkx2.2 consensus sequence, we performed binding competition experiments using unlabeled competitor oligonucleotides and 32P-labeled Nkx2.2 consensus probe, followed by EMSA and PhosphorImager analysis. Table 1 shows the apparent dissociation constants (Kd) for the binding of each of the oligonucleotides to ()Nkx2.2, expressed as a decimal fraction of the apparent Kd for the consensus sequence. Notably, changes involving the core sequence (oligonucleotide M3) can result in as much as a 93% decrease in affinity, and changes involving either or both of the A+T-rich sequences flanking the core (oligonucleotides M2, M4, M5, and M6) result in 30–67% decreases in affinity.
Table 1.
Apparent dissociation constants for binding to ()Nkx2.2
| Oligonucleotide* | Sequence† | Relative binding affinity‡ |
|---|---|---|
| Nkx2.2 consensus | TTTTTAAGTGGTTTTT | 1.0 |
| M1 | TTTTCAAGTGGTTTTT | 5.2 ± 1.0 |
| M2 | GGGTTAAGTGGGGGGG | 0.55 ± 0.17 |
| M3 | TTTTTACGTGGTTTTT | 0.067 ± 0.003 |
| M4 | TTTTTAAGTGGTTGGG | 0.33 ± 0.02 |
| M5 | GGGTTAAGTGGTTTTT | 0.70 ± 0.19 |
| M6 | TTTTTAAGTGGGGTTT | 0.45 ± 0.02 |
Designation for the individual oligonucleotides are the same as those in Fig. 2.
Alignment with the Nkx2.2 consensus sequence is shown. Changes from the consensus are underlined. The complete sequences of the oligonucleotides are provided in Materials and Methods.
Affinities of the individual oligonucleotides for ()Nkx2.2 were determined by competition EMSA, followed by PhosphorImager analysis, as detailed in Materials and Methods. The apparent dissociation constant (Kd) for the consensus oligonucleotide is 3.12 × 10−8 M; the relative affinities of the other oligonucleotides are expressed as a decimal fraction of this value. Data represent a mean ± SEM of at least three separate determinations.
The C Terminus of Nkx2.2 Strongly Activates Transcription.
To determine how Nkx2.2 regulates transcription, we constructed several one-hybrid mammalian expression plasmids with portions of Nkx2.2 fused to the GAL4DBD and tested these in mammalian cotransfection experiments with a reporter plasmid containing five copies of the GAL4 upstream activating sequence (UAS) upstream of either the E1b promoter or the rat prolactin promoter driving a luciferase reporter gene. The mammalian cell lines used in these experiments were the mouse pancreatic β-cell-derived line βTC3 and the fibroblast-derived line NIH 3T3. Western blots and EMSAs using nuclear extracts confirmed that the fusion constructs were expressed at comparable levels and were capable of binding DNA (data not shown).
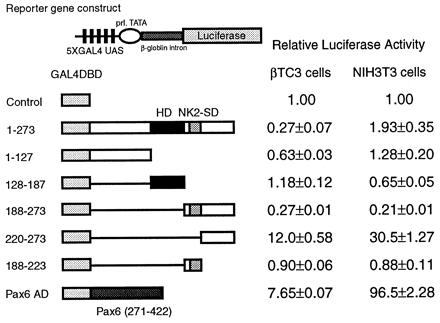
Fig. 2 shows the results of the mammalian cotransfection experiments using the E1b-luciferase reporter, a reporter that is suitable for observing transcriptional activation because of its low basal luciferase activity. The figure demonstrates that all constructs exhibit similar function in the two cell lines. Interestingly, whereas the entire C-terminal region beyond the homeodomain (amino acids 188–273) exhibits no transcriptional activity in isolation, a dramatic increase in transcriptional activity (805-fold in NIH 3T3 cells and 49-fold in β-TC3 cells) is observed when the 33 amino acids containing the NK2-SD are deleted from this C-terminal region. This finding demonstrates that the NK2-SD functionally masks an intrinsic activation domain in the C terminus. Further deletions of the constructs involving the C terminus alone (as shown in Fig. 2C) suggest that the entire C terminus beyond the NK2-SD is necessary for maximum activation.
Figure 2.

The transactivation domain of Nkx2.2 maps to the C terminus. A reporter plasmid containing five tandem copies of the GAL4 UAS upstream of the E1b minimal promoter driving luciferase and an expression plasmid encoding each GAL4DBD-Nkx2.2 fusion construct were cotransfected with a CMV promoter-driven β-galactosidase expression plasmid. All luciferase activities are corrected for β-galactosidase activity. Relative luciferase activities are calculated, with the activity of cells transfected with the GAL4DBD alone set at 1. All data are shown as mean ± SEM. (A) The reporter plasmid is indicated schematically. (B) The results of the one-hybrid assays. (Left) Results from βTC3 cells. (Right) Results from NIH 3T3 cells. (C) Results of detailed mapping of the activation domain. (Left) Results from βTC3 cells. (Right) Results from NIH 3T3 cells.
To identify potential transcriptional repression domains within Nkx2.2, we performed identical mammalian cotransfection experiments, using a reporter vector with high basal activity (prolactin promoter–β-globin intron–luciferase) that allows the detection of repression. As shown in Fig. 3, the full-length protein produces modest, 3- to 4-fold transcriptional repression in βTC3 cells. Whereas this repression activity appears to map to the region encompassing the NK2-SD and C terminus (amino acids 188–273), neither the NK2-SD (amino acids 188–223) nor the C terminus (amino acids 220–273) by itself demonstrates transcriptional repression. This result implies that the ability of the NK2-SD to mask the activation potential of the C terminus is not itself a result of intrinsic repression activity.
Figure 3.
Full-length Nkx2.2 acts as a weak repressor in βTC3 cells. A reporter plasmid containing five tandem copies of the GAL4 UAS upstream of the prolactin minimal promoter driving luciferase and an expression plasmid encoding each GAL4DBD-Nkx2.2 fusion construct or a GAL4DBD-Pax6 activation domain fusion construct were cotransfected with CMVβgal. All luciferase activities are corrected for β-galactosidase activity. Relative luciferase activities are calculated, with the activity of cells transfected with the GAL4DBD alone set at 1. All data are shown as mean ± SEM.
The Hydrophobic Character of the NK2-SD Negatively Regulates Transcriptional Activation.
As noted above, the NK2-SD of Nkx2.2 appears to shield a potentially powerful activation domain in the native molecule. The NK2-SD is a highly conserved motif that defines members of the NK-2 family of transcription factors in both mammals and invertebrates. The sequence of the NK2-SD of Nkx2.2, as shown in Fig. 4, consists of a well-conserved hydrophobic core region (VAVPVLV) and a conserved serine at the N terminus (PLPSP) that resembles a potential mitogen-activated protein kinase phosphorylation site, PXnTSP (20). To assess whether the hydrophobic region or potential phosphorylation site within the NK2-SD might contribute to the observed inhibition of transactivation, we generated two mutant Nkx2.2 constructs and tested them in our mammalian one-hybrid cotransfection assay. Mutation S199A (Ala-for-Ser mutation at position 199) disrupts the potential mitogen-activated protein kinase site, whereas mutation A204E, L208E (Glu-for-Ala at position 204 and Glu-for-Leu at position 208) disrupts local hydrophobicity (Fig. 4A). As shown in Fig. 4 C and D, when mutation S199A is introduced into either the full-length or the N-terminally truncated protein, no effect on transactivation is observed. In contrast, when mutation A204E, L208E is introduced into the same constructs, significant increases in transactivation are observed. These results suggest that the hydrophobic character of NK2-SD is essential for functionally masking the activation domain.
Figure 4.
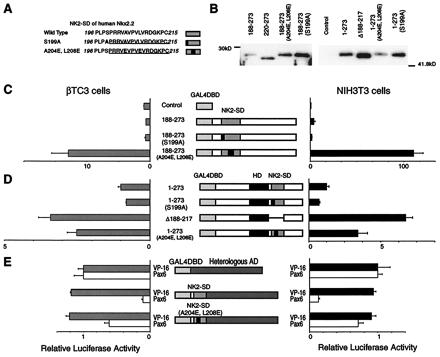
Characterization of the transactivation domain of Nkx2.2. A reporter plasmid containing five tandem copies of the GAL4 UAS upstream of the E1b minimal promoter driving luciferase and an expression plasmid encoding each GAL4DBD-Nkx2.2 fusion construct were cotransfected with a CMV promoter-driven β-galactosidase expression plasmid. All luciferase activities are corrected for β-galactosidase activity. All data are shown as mean ± SEM. (A) The sequences of the native and mutant NK2-SD. The NK2-SD is underlined, and mutations are in boldface. (B) Western blotting data. Each construct is transfected into NIH 3T3 cells. Five micrograms of nuclear extracts from each cell is loaded. (C) The relative luciferase activities from βTC3 cells and NIH 3T3 cells transfected with expression plasmids encoding the C-terminal region with wild-type or mutant NK2-SD fused to the GAL4DBD. Relative luciferase activities are calculated, with the activity of cells transfected with the GAL4DBD alone set at 1. (D) The relative luciferase activities from βTC3 cells and NIH 3T3 cells transfected with expression plasmids encoding the full-length Nkx2.2 with wild-type or mutant NK2-SD fused to the GAL4DBD. Relative luciferase activities are calculated, with the activity of cells transfected with GAL4DBD–full-length Nkx2.2 alone set at 1. Comparison with the GAL4DBD alone is shown in Fig. 2B. (E) Relative luciferase activities from cells transfected with the VP16 activation domain or Pax6 activation domain fused to wild-type or mutant NK2-SD and the GAL4DBD. Relative luciferase activities are calculated, with the activity of cells transfected with an expression plasmid containing the isolated VP16 activation domain fused to the GAL4DBD set at 1. VP16 activation domain fused to the GAL4DBD showed 516,000-fold activation in NIH 3T3 cells and 5,990-fold activation in βTC3 cells compared with the GAL4DBD alone. On the other hand, Pax6 activation domain fused to the GAL4DBD showed 102-fold activation in NIH 3T3 cells and 6.1-fold activation in βTC3 cells compared with the GAL4DBD alone.
The NK2-SD Masks the Activity of a Heterologous Activation Domain and Cannot Function in Trans.
To determine whether the NK2-SD can function as a general inhibitor of transcriptional activation domains, we fused the NK2-SD to heterologous activation domains and tested the ability of the fusion proteins to activate transcription, using the E1b-luciferase reporter. As shown in Fig. 4E, the NK2-SD does not inhibit the transcriptional activation caused by the powerful viral VP16 activation domain, but it does inhibit the activity of the weaker activation domain from the paired homeodomain factor Pax6.
It is possible that the observed inhibition of activation by the NK2-SD is a result of a direct interaction between the NK2-SD and the activation domain of Nkx2.2. In this case, we would expect that coexpression of the NK2-SD (amino acids 188–223) with the yeast GAL4DBD–Nkx2.2 C-terminal (amino acids 220–273) fusion should mitigate the transcriptional activation caused by the fusion protein alone. However, we did not observe inhibition by the NK2-SD in this experiment (data not shown).
The NK2-SD Functionally Masks the C-Terminal Activation Domain in the Context of the Native Nkx2.2 Molecule.
To determine whether the NK2-SD and activation domain function in the native form of Nkx2.2 (as opposed to the GAL4 one-hybrid fusion), we coexpressed various portions of Nkx2.2 with a reporter vector containing seven copies of the Nkx2.2 consensus binding site upstream of prolactin-luciferase. As shown in Fig. 5, whereas most Nkx2.2 constructs demonstrated no significant transcriptional activity in our reporter assay, it is notable that the construct with the NK2-SD deleted was able to activate transcription significantly over baseline, confirming our observation in the one-hybrid experiments. The NK2-SD also inhibits the activity of full-length Nkx2.2 in βTC3 cells (data not shown).
Figure 5.
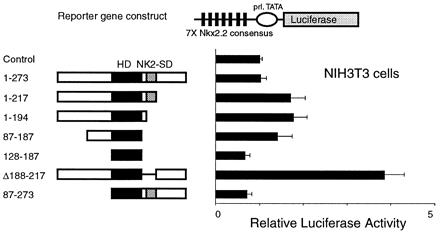
Removal of the NK2-SD activates Nkx2.2. A reporter plasmid containing seven tandem copies of the Nkx2.2 DNA-binding consensus site (shown in Table 1) upstream of the prolactin promoter driving luciferase (pFOxLuc1–7XNk2) was cotransfected with the CMV promoter-driven expression plasmids encoding the Nkx2.2 cDNA shown. Relative luciferase activities are calculated, with the activity of cells transfected with a CMV expression plasmid without cDNA insert set at 1. All data are shown as mean ± SEM.
Discussion
The NK2-SD is a highly conserved motif that is found in all vertebrate NK-2 class transcription factors and the Drosophila NK2 transcription factor (1); this fact suggests a potentially important role for this domain in transcription factor function. Our studies addressed the possible role of the NK2-SD in the DNA-binding and transactivation properties of Nkx2.2. We demonstrate that ()Nkx2.2 (a construct encompassing the homeodomain and NK2-SD) recognizes the DNA sequence 5′-TCTAAGTGAGCTT-3′; this sequence contains the central core sequence TCTAAG, which is consistent with the known binding cores for other NK2 proteins (7, 9). A similar PCR-based oligonucleotide selection technique was used by Chen et al. (9) to identify the DNA-binding sequence for the homeodomain of Nkx2.5. Although they obtained a similar binding sequence using just the homeodomain of Nkx2.5 (5′-TNAAGTG-3′), we note two significant differences between their findings and ours: (i) Chen et al. observed an additional weak binding sequence containing a TAAT core, whereas we did not for Nkx2.2, and (ii) whereas the binding sequence for Nkx2.5 contained only 7 bp (TNAAGTG), we found that ()Nkx2.2 binds a highly reproducible 10-bp sequence that is flanked on both ends by a stretch of A+T-rich sequences (Fig. 1). Although the presence of the NK2-SD in our protein construct might contribute to these observed differences, we believe that these differences most likely derive from the greater number of cycles of selection we used (eight versus four). Indeed, the fact that the isolated homeodomain of Nkx2.2 and ()Nkx2.2 display parallel affinities for changes in the consensus sequence demonstrates that the NK2-SD contributes little to overall DNA binding specificity.
In mammalian cell transcription assays, full-length Nkx2.1 and Nkx2.5 have been shown to activate reporter gene transcription (9, 10). We show here, however, that full-length Nkx2.2 produces no significant activation in any of the cell lines studied; in fact, only weak repression activity was noted in the β cell line βTC3. Our detailed mapping studies of potential functional domains in Nkx2.2 reveal that there is a strong activation domain within the C terminus (amino acids 220–273), but that the NK2-SD in the full-length protein functionally masks this activation function. Previous mapping studies with Nkx2.1 and Nkx2.5 are consistent with our conclusion, because the deletion of a region encompassing the NK2-SD significantly increases the activation potential of both of these transcription factors (9, 10). The masking or attenuation of transactivation domains by other portions of the same molecule has been described for transcription factors ATF-2, c-Myb, and Lmx1.1/1.2 (21–23). For ATF-2, its DNA-binding domain has been implicated in this role and was shown to function by interacting directly with its activation domain. In contrast, the NK2-SD of Nkx2.2 does not appear to physically interact with the activation domain, because it does not inhibit activation in trans. Thus it is possible that the NK2-SD, perhaps in conjunction with a cofactor, propagates a conformational change that destabilizes the activation domain.
Within the framework of an intact promoter, multiple factors interact with each other and with promoter elements to regulate transcriptional activity. In the case of NK-2 family members, Nkx2.5 is known to physically interact with GATA4, another transcription factor expressed early in cardiac development, to synergistically activate its target genes (24, 25). Similarly, interaction with Nkx2.2 through its NK2-SD might provide cell type-specific expression of particular target genes. In this regard, the stretch of hydrophobic residues within the NK2-SD is reminiscent of motifs that are known to be interfaces for protein–protein interactions in other DNA-binding factors (8). Accordingly, a mutation (A204E, L208E) that disrupts local hydrophobicity in the NK2-SD restores the ability to activate transcription.
Taken together, our data suggest that the highly conserved NK2-SD functions to control the transactivation properties of NK-2 family members. Although we studied the developmentally important transcription factor Nkx2.2 as a model for elucidating the role of the NK2-SD, retrospective analysis of previous studies (9, 10) suggests that the NK2-SD functions similarly in the related factors Nkx2.1 and Nkx2.5. Furthermore, our studies provide a framework for identifying target genes for Nkx2.2 and understanding the manner in which these target genes are regulated and how alterations of Nkx2.2 function may contribute to genetic forms of diabetes (14, 17). Therefore, our results emphasize the complexity of transcription factor regulation: whereas transcription factors recognize and bind to specific DNA sequences and thereby achieve genetic specificity, that specificity can be modified further by structural and functional effects of specific sequences within the molecule and by the interaction with transcription factors that bind to neighboring sequences.
Acknowledgments
We thank Graeme I. Bell and Hiroto Furuta for kindly providing the human Nkx2.2 gene. We thank Joey Leung for excellent technical assistance and members of the German laboratory for helpful comments and criticisms. H.W. is a recipient of a Juvenile Diabetes Foundation International Postdoctoral Fellowship, and R.G.M. is a recipient of a Research Career Award (K08) from the National Institutes of Health. This work was supported by National Institutes of Health Grant DK21344.
Abbreviations
- TN domain
tin domain
- NK2-SD
NK2-specific domain
- EMSA
electrophoretic mobility-shift assay
- GAL4DBD
yeast GAL4 DNA-binding domain
- UAS
upstream activating sequence
- CMV
cytomegalovirus
References
- 1.Harvey R P. Dev Biol. 1996;178:203–216. doi: 10.1006/dbio.1996.0212. [DOI] [PubMed] [Google Scholar]
- 2.Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox C H, Ward J M, Gonzalez F J. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- 3.Sussel L, Marin O, Kimura S, Rubenstein J L. Development (Cambridge, UK) 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 4.Lints T J, Parsons L M, Hartley L, Lyons I, Harvey R P. Development (Cambridge, UK) 1993;119:419–431. doi: 10.1242/dev.119.2.419. [DOI] [PubMed] [Google Scholar]
- 5.Biben C, Harvey R P. Genes Dev. 1997;11:1357–1369. doi: 10.1101/gad.11.11.1357. [DOI] [PubMed] [Google Scholar]
- 6.Tsao D H, Gruschus J M, Wang L H, Nirenberg M, Ferretti J A. J Mol Biol. 1995;251:297–307. doi: 10.1006/jmbi.1995.0435. [DOI] [PubMed] [Google Scholar]
- 7.Tsao D H, Gruschus J M, Wang L H, Nirenberg M, Ferretti J A. Biochemistry. 1994;33:15053–15060. doi: 10.1021/bi00254a014. [DOI] [PubMed] [Google Scholar]
- 8.Apergis G A, Crawford N, Ghosh D, Steppan C M, Vorachek W R, Wen P, Locker J. J Biol Chem. 1998;273:2917–2925. doi: 10.1074/jbc.273.5.2917. [DOI] [PubMed] [Google Scholar]
- 9.Chen C Y, Schwartz R J. J Biol Chem. 1995;270:15628–15633. doi: 10.1074/jbc.270.26.15628. [DOI] [PubMed] [Google Scholar]
- 10.De Felice M, Damante G, Zannini M, Francis-Lang H, Di Lauro R. J Biol Chem. 1995;270:26649–26656. doi: 10.1074/jbc.270.44.26649. [DOI] [PubMed] [Google Scholar]
- 11.Price M, Lazzaro D, Pohl T, Mattei M G, Ruther U, Olivo J C, Duboule D, Di Lauro R. Neuron. 1992;8:241–255. doi: 10.1016/0896-6273(92)90291-k. [DOI] [PubMed] [Google Scholar]
- 12.Rudnick A, Ling T Y, Odagiri H, Rutter W J, German M S. Proc Natl Acad Sci USA. 1994;91:12203–12207. doi: 10.1073/pnas.91.25.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briscoe J, Sussel L, Serup P, Hartigan-O'Connor D, Jessell T M, Rubenstein J L, Ericson J. Nature (London) 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- 14.Sussel L, Kalamaras J, Hartigan-O'Connor D J, Meneses J J, Pedersen R A, Rubenstein J L, German M S. Development (Cambridge, UK) 1998;125:2213–2221. doi: 10.1242/dev.125.12.2213. [DOI] [PubMed] [Google Scholar]
- 15.Catron K M, Iler N, Abate C. Mol Cell Biol. 1993;13:2354–2365. doi: 10.1128/mcb.13.4.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epstein J, Cai J, Glaser T, Jepeal L, Maas R. J Biol Chem. 1994;269:8355–8361. [PubMed] [Google Scholar]
- 17.Furuta H, Horikawa Y, Iwasaki N, Hara M, Sussel L, Le Beau M M, Davis E M, Ogata M, Iwamoto Y, German M S, Bell G I. Diabetes. 1998;47:1356–1358. doi: 10.2337/diab.47.8.1356. [DOI] [PubMed] [Google Scholar]
- 18.Smith S B, Ee H C, Conners J R, German M S. Mol Cell Biol. 1999;19:8272–8280. doi: 10.1128/mcb.19.12.8272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohneda K, Mirmira R G, Wang J, Johnson J D, German M S. Mol Cell Biol. 2000;20:900–911. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez E, Northwood I C, Gonzalez F A, Latour D A, Seth A, Abate C, Curran T, Davis R J. J Biol Chem. 1991;266:15277–15285. [PubMed] [Google Scholar]
- 21.Johnson J D, Zhang W, Rudnick A, Rutter W J, German M S. Mol Cell Biol. 1997;17:3488–3496. doi: 10.1128/mcb.17.7.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X Y, Green M R. Genes Dev. 1996;10:517–527. doi: 10.1101/gad.10.5.517. [DOI] [PubMed] [Google Scholar]
- 23.Dash A B, Orrico F C, Ness S A. Genes Dev. 1996;10:1858–1869. doi: 10.1101/gad.10.15.1858. [DOI] [PubMed] [Google Scholar]
- 24.Searcy R D, Vincent E B, Liberatore C M, Yutzey K E. Development (Cambridge, UK) 1998;125:4461–4470. doi: 10.1242/dev.125.22.4461. [DOI] [PubMed] [Google Scholar]
- 25.Sepulveda J L, Belaguli N, Nigam V, Chen C Y, Nemer M, Schwartz R J. Mol Cell Biol. 1998;18:3405–3415. doi: 10.1128/mcb.18.6.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]