

Abstract
Vanillyl-alcohol oxidase (VAO) is the prototype of a newly recognized family of structurally related oxidoreductases sharing a conserved FAD-binding domain. The active site of VAO is formed by a cavity where the enzyme is able to catalyze many reactions with phenolic substrates. Among these reactions is the stereospecific hydroxylation of 4-ethylphenol-forming (R)-1-(4′-hydroxyphenyl)ethanol. During this conversion, Asp-170 is probably critical for the hydration of the initially formed p-quinone methide intermediate. By site-directed mutagenesis, the putative active site base has been relocated to the opposite face of the active site cavity. In this way, a change in stereospecificity has been achieved. Like native VAO, the single mutants T457E, D170A, and D170S preferentially converted 4-ethylphenol to the (R)-enantiomer of 1-(4′-hydroxyphenyl)ethanol. The double mutants D170A/T457E and D170S/T457E exhibited an inverted stereospecificity with 4-ethylphenol. Particularly, D170S/T457E was strongly (S)-selective, with an enantiomeric excess of 80%. The crystal structure of D170S/T457E, in complex with trifluoromethylphenol, showed a highly conserved mode of ligand binding and revealed that the distinctive catalytic properties of this mutant are not caused by major structural changes.
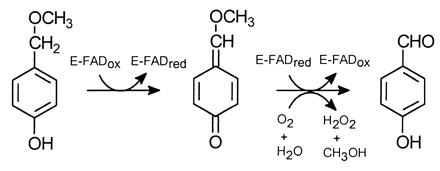
Vanillyl-alcohol oxidase (VAO; EC 1.1.3.38) is a flavoenzyme from Penicillium simplicissimum containing a covalently bound FAD. The enzyme is the prototype of a newly recognized family of structurally related oxidoreductases sharing a conserved FAD-binding domain (1). Prominent members of this family include MurB (UDP-N-acetylenolpyruvylglucosamine reductase) (2), the molybdenum enzyme carbon monoxide dehydrogenase (3), and the flavocytochrome p-cresol methylhydroxylase (PCMH) (4). VAO is active with a wide range of phenolic compounds (5, 6). Conversion of the physiological substrate 4-(methoxymethyl)phenol by VAO involves the initial transfer of a hydride from the substrate to the flavin, resulting in the formation of a complex between the two-electron reduced enzyme and the p-quinone methide product intermediate. Next, the reduced flavin is reoxidized by molecular oxygen with the concomitant hydration of the p-quinone methide (7), as shown in Scheme S1:
Scheme 1.
The crystal structure of VAO represents the first crystal structure of a flavoenzyme with a histidyl-bound flavin (8). Each VAO monomer comprises two domains, with the cap domain covering the active site cavity and the larger domain forming the binding site of the FAD prosthetic group. All present evidence suggests that the covalent anchoring of the flavin is a self-catalytic process (9). From the properties of His-422 variants, it was established that the covalent interdomain interaction with His-422 serves to increase the flavin redox potential, an essential feature for efficient redox catalysis (10). Another important residue in the enzyme active site is Asp-170. The side chain of this acidic residue is only 3.6 Å from flavin N5 and the reactive methylene group of the phenolic substrate (8). From studies on Asp-170 variants, we recently showed that Asp-170 is important for maintaining the high redox potential of the enzyme and thus its reactivity (11). Furthermore, these studies revealed that Asp-170 assists in covalent flavinylation, possibly by donating a proton to flavin N5 (11). It has been suggested that, in the related flavocytochrome PCMH, Glu-380 fulfills a similar role during the flavinylation process (4, 11, 12).
VAO and PCMH have a similar substrate specificity (6, 13, 14), and the active site architecture of both enzymes is highly conserved (4, 8). However, VAO preferentially converts 4-ethylphenol and 4-propylphenol to the corresponding (R)-1-(4′-hydroxyphenyl)alcohols (14), whereas PCMH favors the production of the (S)-enantiomers (15, 16). The structural determinants involved in the stereochemistry of VAO and PCMH are unknown. It has been proposed that Asp-170 in VAO (8, 11) and the equivalent Glu-380 in PCMH (4) might activate the water involved in substrate conversion. However, in PCMH another acidic residue (Glu-427) is situated near the reactive carbon of bound p-cresol at the opposite face of the substrate (4). In VAO, this position is occupied by Thr-457 (Fig. 1), raising the possibility that this structural variation plays a role in the opposite stereospecificity of water attack.
Figure 1.
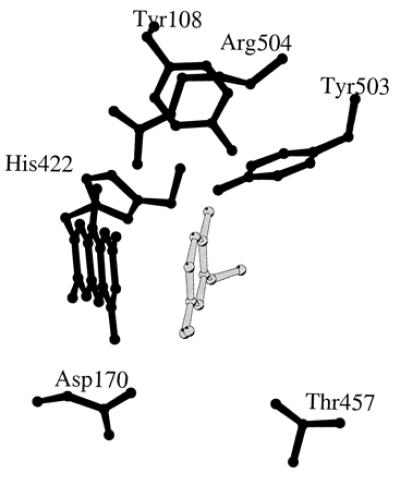
Drawing of the active site cavity of isoeugenol-complexed VAO. The side chains of Asp-170 and Thr-457 in VAO align with Glu-380 and Glu-427 in PCMH, respectively. This figure was prepared with molscript (17).
To investigate this hypothesis and the function of Asp-170 in the active site of VAO in further detail, we have generated the single mutant T457E and the double mutants D170A/T457E and D70S/T457E. Here, we present the biochemical and structural characterization of the first VAO variant with an inverted stereospecificity. Furthermore, evidence is provided that Glu-457 may substitute for Asp-170 in promoting the covalent tethering of the flavin ring.
Materials and Methods
Materials.
Escherichia coli strain DH5αF′ (18) and the plasmid pUCBM20 (Boehringer Mannheim) were used for cloning throughout, whereas E. coli strain TG2 (19) and the plasmid pEMBL19(−) (Boehringer Mannheim) were used for expression of the vaoA gene. Yeast extract and tryptone peptone extract were from Difco. Racemic 1-(4′-hydroxyphenyl)ethanol was synthesized as described (14), and (R)-1-(4′-hydroxyphenyl)ethanol was obtained from the enzymatic conversion of 4-ethylphenol. All other chemicals were purchased as reported earlier (6, 11).
Site-Directed Mutagenesis and Enzyme Purification.
pBC11 (wild-type), pBC15 (D170S), and pBC16 (D170A) were constructed as described before (11). The SalI-KpnI fragment of pBC11 was ligated in pUCBM20 and used for PCR-based mutagenesis with the oligonucleotide 5′-GATTTTATCGGCGAGTTCACAGTCGGTATG-3′ (where GAG denotes the replacement of Thr by Glu). The mutated SalI–KpnI fragment was ligated into pBC11, pBC15, and pBC16, yielding pBC22 (T457E), pBC24 (D170S/T457E), and pBC23 (D170A/T457E), respectively. Successful mutagenesis was confirmed by plasmid sequencing. Transformed E. coli cells were grown in Luria-Bertani medium supplemented with 75 μg/ml ampicillin and 0.25 mM isopropyl β-d-thiogalactopyranoside (20). The VAO mutant proteins were purified according to a previously established procedure (11). Enzyme purity was checked by SDS/PAGE and by analytical gel filtration using a Superdex (Amersham Pharmacia) 200 HR 10/30 column.
Analytical Methods.
All experiments were performed in air-saturated 50 mM potassium phosphate buffer (pH 7.5) at 25°C, unless stated otherwise. Molar absorption coefficients for protein-bound flavin and the fraction of covalently bound flavin were determined as described previously (11). SDS/PAGE was carried out in 12.5% slab gels essentially as reported earlier (21, 22). The redox potentials of the VAO mutants were determined by the xanthine/xanthine oxidase method (23), essentially as described before (10, 11).
HPLC experiments were performed with an Applied Biosystems pump equipped with a Waters 996 photodiode array detector and a 3.9 × 150-mm Waters Novapak C18 column, essentially as described earlier (6). GC analysis was performed on a Varian Star 3400 CX gas chromatograph with a 50-m capillary Chrompack CP-cyclodextrin column (0.25-mm internal diameter and 0.25-μM film thickness) having CP-cyclodextrin-B-2,3,6-M-19 as the chiral stationary phase. The temperature program started at 80°C, followed by an increase with 7°C min−1 to 160°C, which was held for 30 min (14).
VAO activity was determined by following absorption spectral changes of aromatic substrates or by measuring the rate of product formation by HPLC (6, 11). Rapid reaction stopped-flow kinetics were conducted with a Hi-Tech SF-51 apparatus equipped with a Hi-Tech M300 monochromator diode-array detector (Salisbury, U.K.), essentially as described (7). Deconvolution of spectral data were performed by using the specfit global analysis program version 2.10 (Spectrum Software Associates, Chapel Hill, NC).
Crystallographic Analysis of Mutant D170S/T457E.
Crystals of the double mutant D170S/T457E were grown by using the hanging-drop vapor diffusion method, in conditions similar to those for wild-type VAO (8). Briefly, protein solutions containing 10 mg protein per ml in 50 mM potassium phosphate buffer (pH 7.5) were equilibrated against a reservoir solution containing 100 mM sodium acetate/hydrochloride (pH 4.6) and 3% (wt/vol) PEG 4000. Binding of the inhibitor trifluoromethylphenol was achieved by soaking the crystals for 5 h in a solution containing 10 mM of the substrate analog. Diffraction data were measured from a single crystal at the X11 beam line of the European Molecular Biology Laboratory/Deutsches Elektronen Synchrotron (Hamburg, Germany) by using a MarCCD detector at 100 K. Before data collection, the crystal was exposed for a few seconds to a cryoprotecting solution containing 20% (vol/vol) PEG 400/10% (wt/vol) PEG 4000/50 mM sodium acetate/hydrochloride (pH 4.6). The data were processed by using mosflm (24) and programs of the ccp4 package (25). The double mutant crystals are isomorphous to those of wild-type enzyme and belong to space group I4 with unit cell parameters a = b = 131.28, c = 134.36 Å. Crystallographic refinement was performed with the refmac program (26). A bulk solvent correction was applied by using the programs of the ccp4 package whereas the positions of ordered solvent molecules were located by using arp (27). The free R-factor was used to monitor the progress of the refinement. Electron density maps were visually inspected by using the program o (28). See Table 2 for a summary of the final refinement statistics.
Table 2.
Data collection and refinement statistics for T457E/D170S VAO in complex with trifluoromethylphenol
| Measurement | Value |
|---|---|
| Resolution, Å | 20–2.75 |
| Observed reflections | 69,063 |
| Unique reflections | 28,740 |
| Completeness of data, %* | 97.3 (95.0) |
| Multiplicity* | 2.4 (2.2) |
| Intensities I/σ* | 7.4 (2.1) |
| Rsym, %*† | 11.0 (37.1) |
| Cell dimensions, Å | a = b = 131.28, c = 134.36 |
| Rfactor, % | 22.5 |
| Rfree, % | 28.7 |
| No. of ligand atoms | 11 (trifluoromethylphenol) |
| rmsd for ideal value‡ | |
| Bond lengths, Å | 0.009 |
| Bond angles, ° | 2.5 |
| Trigonal atoms, Å | 0.019 |
| Planar groups, Å | 0.001 |
The values relating to the highest resolution shell (2.90–2.75 Å) are given in brackets.
Rsym = ∑|Ij − 〈Ij〉|/∑〈Ij〉, where Ij is the intensity of an observation of reflection j and 〈Ij〉 is the average intensity for reflection j.
The rms deviations (rmsd) were calculated using the program refmac (26).
Results
Enzyme Characterization.
The purified VAO mutants T457E, D170A/T457E, and D170S/T457E were bright yellow, and their near UV absorption properties were virtually identical to wild-type enzyme (29). Fluorescence analysis of unstained SDS gels showed that the mutant enzymes were as fluorescent as wild-type VAO. This fluorescence behavior, and the fact that the mutants did not release any flavin upon trichloroacetic acid treatment (29), indicated that all flavin was covalently bound. Moreover, analytical gel filtration revealed that the mutants were mainly octameric, with only a small fraction in the dimeric form. This oligomerization behavior is similar to wild-type VAO (30, 31).
The redox potentials of the VAO mutants were measured by the xanthine/xanthine oxidase system (23) by using the reference dyes thionin (Em = +60 mV) and methylene blue (Em = +11 mV). As found for wild-type enzyme (10), all three mutants were reduced in a single two-electron step, with no formation of any semiquinone (Fig. 2). On plotting log (Eox/Ered) vs. log (dyeox/dyered) (32, 33), midpoint redox potentials of +20 ± 1 mV, +22 ± 2 mV, and +31 ± 2 mV were estimated for T457E, D170A/T457E, and D170S/T457E, respectively. These values are lower than for wild-type VAO (Em = +55 mV) (10), but considerably higher than the value reported for D170S (Em = −91 mV) (11).
Figure 2.
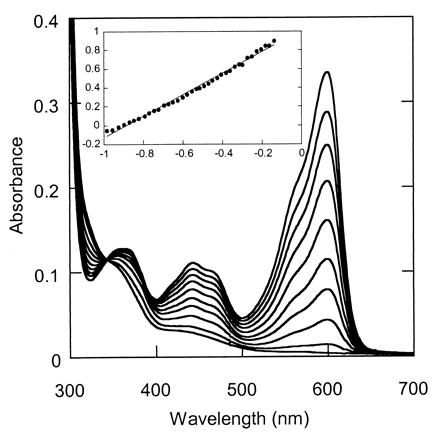
Anaerobic reduction of 8 μM D170S/T457E by the xanthine/xanthine oxidase system (23) in the presence of 8 μM thionin in 50 mM potassium phosphate buffer (pH 7.5) at 25°C. Spectra were taken at regular time intervals (10 min), and full reduction was achieved after 95 min. The Inset shows the log (Eox/Ered) (measured at 440 nm after correction for thionin) vs. log (dyeox/dyered) (measured at 600 nm) plot.
Catalytic Properties.
VAO converts 4-(methoxymethyl)phenol with a maximum turnover rate of 3.1 s−1 and a Km value of 55 μM (7). Replacement of Thr-457 by Glu decreased the catalytic efficiency (kcat/Km) with this substrate by about a factor of 7 (Table 1). The double mutants D170A/T457E and D170S/T457E were rather poor catalysts (Table 1). Nevertheless, these mutants were considerably more active with 4-(methoxymethyl)phenol than D170A and D170S (11).
Table 1.
Kinetic parameters of wild type VAO and Asp-170/Thr-457 mutants with 4-(methoxymethyl)phenol in air-saturated 50 mM potassium phosphate buffer (pH 7.5) at 25°C
Previous studies have shown that the turnover rate of VAO with 4-(methoxymethyl)phenol is mainly determined by the rate of flavin reduction. Moreover, rapid kinetics have established that, in the absence of oxygen, the reduced enzyme forms a rather stable complex with the p-quinone methide intermediate of the substrate (7), shown in Scheme 2:
 |
Scheme 2.
In analogy to wild-type enzyme, anaerobic reduction of T457E by 4-(methoxymethyl)phenol followed monoexponential kinetics, resulting in fully reduced enzyme when monitored at 440 nm. The maximum reduction rate of 1.2 s−1 approached the maximum turnover rate (Table 1), indicating that flavin reduction mainly determines the rate of overall catalysis. Diode-array spectral analysis of the anaerobic reduction of T457E with 4-(methoxymethyl)phenol revealed the formation of a strong absorption band with a maximum at 364 nm, representing the binary complex between the reduced enzyme and the p-quinone methide product intermediate. As in wild-type VAO (11), the decomposition of this complex, forming 4-hydroxybenzaldehyde, was very slow (kobs = 0.01 s−1).
The rate of anaerobic reduction of D170A/T457E and D170S/T457E by 4-(methoxymethyl)phenol was 0.02 s−1 and 0.03 s−1, respectively. Again, these values are in the same range as the corresponding turnover rates (Table 1). With both double mutants, the flavin became fully reduced in a monophasic process with the concomitant formation of 4-hydroxybenzaldehyde. In contrast to T457E and wild-type VAO, no stabilization of the p-quinone methide-reduced enzyme complex was observed. A similar behavior was recently observed for D170A and D170S (11).
Stereochemistry of the Mutant Enzymes.
Wild-type VAO hydroxylates 4-ethylphenol stereospecifically to (R)-1-(4′-hydroxyphenyl)ethanol, with an enantiomeric excess of 94% (14). To probe the role of Asp-170 and Thr-457 in this reaction, we studied the conversion of 4-ethylphenol by the single mutants T457E, D170A, and D170S and by the double mutants D170A/T457E and D170S/T457E. The turnover rates of the mutant enzymes with 4-ethylphenol were about two orders of magnitude lower than with wild-type VAO (not shown). Nevertheless, all mutants catalyzed the formation of 1-(4′-hydroxyphenyl)ethanol in high yield. As found for native VAO (14), only minor amounts of the side product 1-(4′-hydroxyphenyl)ethene were formed. T457E showed a high preference for the formation of the (R)-enantiomer of 1-(4′-hydroxyphenyl)ethanol (Fig. 3A). D170A and D170S were less (R)-selective, with enantiomeric excess values of 26% and 50%, respectively. The double mutants exhibited an opposite stereospecificity with 4-ethylphenol. Especially, D170S/T457E showed a strong preference for the (S)-enantiomer, with an enantiomeric excess of 80% (Fig. 3B).
Figure 3.
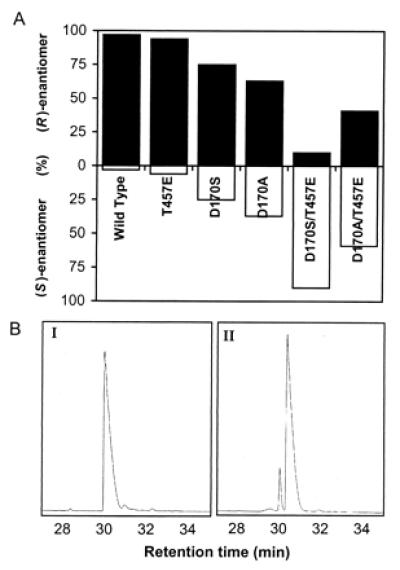
(A) Stereospecificity of hydroxylation of 4-ethylphenol by wild-type VAO and Asp-170/Thr-457 mutants in 50 mM potassium phosphate buffer (pH 7.5) at 25°C. (R)-1-(4′-hydroxyphenyl)ethanol (black) and (S)-1-(4′-hydroxyphenyl)ethanol (white). (B) Expanded chiral GC separation pattern of the hydroxylation of 4-ethylphenol by (I) wild-type VAO and (II) D170S/T457E.
Structure of D170S/T457E.
The crystal structure of D170S/T457E in complex with the substrate analog trifluoromethylphenol was determined at 2.75 Å resolution (Table 2). The inhibitor was clearly visible in the electron density map of one of the two crystallographically independent subunits (subunit B). Its orientation in the proximity of the flavin ring was nearly identical to the orientation in several previously determined enzyme–inhibitor complex structures (8, 10, 11). Superposition of the structures of trifluoromethylphenol-complexed D170S/T457E and isoeugenol-complexed wild-type VAO (8) showed that the two amino acid replacements did not cause any significant conformational change, which is reflected in a rms deviation for all Cα-atoms of only 0.23 Å. The active site cavity of D170S/T457E was highly similar to that of wild-type enzyme (8) (Fig. 4) and D170S (11). The Oγ-atom of Ser-170 in D170S/T457E points toward flavin N5 at a distance of 3.7 Å from the isoalloxazine ring. Compared with Ser-170, the newly introduced Glu-457 is positioned at the opposite side of the substrate-binding cavity. Its side chain points toward the Cα-atom of bound trifluoromethylphenol, resulting in a distance between the carboxylic moiety of Glu-457 and the Cα-atom of the inhibitor of 3.5 Å. In wild-type VAO, Thr-457 points also toward this Cα-atom; however, the distance between the inhibitor Cα and Oγ of Thr-457 is 6.2 Å (8).
Figure 4.
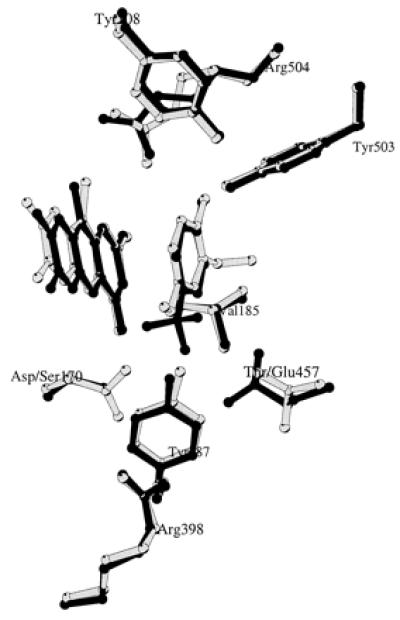
Drawing comparing the active site cavities of trifluoromethylphenol-complexed D170S/T457E (black) and isoeugenol-complexed wild-type VAO (gray). This figure was prepared with molscript (17).
Discussion
The active site topology of VAO is highly similar to that of the flavocytochrome PCMH(4, 8). Nevertheless, subtle structural differences between both flavoenzymes exist that are likely of relevance with respect to the mechanism and specificity of catalysis. One striking difference is the arrangement of acidic residues (Fig. 1). The active site of VAO contains a single aspartate (Asp-170) whose side chain is close to flavin N5 and the reactive Cα-atom of the substrate (8). In contrast, PCMH harbors two glutamates at opposite faces of the substrate (4). From this location, we rationalized that the different arrangement of acidic residues might explain the opposite stereochemistry in the reaction with 4-alkylphenols (14, 15, 16).
The spectral and hydrodynamic properties of T457E, D170A/T457E, and D170S/T457E were similar to wild-type VAO. Moreover, the crystal structure of D170S/T457E established that the introduction of an acidic side chain at position 457 does not result in any significant structural perturbation. In all three mutants, and different from D170A and D170S (11), the flavin prosthetic group was fully covalently bound.
Previous studies have indicated that the covalent interaction between His-422 and the isoalloxazine ring markedly increases the redox potential of the flavin cofactor (10). The redox potentials of D170A/T457E and D170S/T457E were only slightly lower than that of wild-type VAO, supporting the proposal that an acidic residue in the active site cavity is required for maintaining the high redox potential (11). Self-catalytic covalent flavinylation is thought to occur via initial flavin tautomerization (34, 35), followed by donation of a proton to the iminoquinone methide form of the flavin (12). In VAO, Asp-170 is envisaged to act as the proton donor in this process (11). Because D170A/T457E and D170S/T457E contained fully covalently bound flavin, this indicates that Glu-457 can replace Asp-170 in the process of covalent flavinylation. The structure of D170S/T457E showed that the carboxylic moiety of Glu-457 is about 7.0 Å from flavin N5. This distance suggests that Glu-457 cannot donate a proton directly to the flavin and that the proton might be transferred via a water molecule, as observed in sarcosine oxidase (12), rather than by direct donation from Glu-457.
The single mutant T457E was rather active with 4-(methoxymethylphenol) and fully capable of stabilizing the complex between the reduced enzyme and the p-quinone methide intermediate. From this catalytic performance and the relatively high redox potential of T457E, we conclude that the Thr-457-Glu replacement has no profound effects on the electrostatic interactions in the enzyme active site. The double mutants reacted more slowly with 4-(methoxymethylphenol) and did not stabilize the quinone methide. This behavior strongly supports our earlier proposal that Asp-170 is essential for efficient redox catalysis by forming a hydrogen bond with the protonated N5-atom of the reduced flavin (11).
Wild-type VAO preferentially converts 4-ethylphenol to the (R)-enantiomer of 1-(4′-hydroxyphenyl)ethanol (14). The high yield of this asymmetric compound results from the stereospecific attack of a water molecule to the p-quinone methide intermediate in the active site. The D170A/T457E and D170S/T457E double mutants exhibit an inverted stereospecificity with 4-ethylphenol. This (S)-selectivity is likely caused by the attack of a water molecule from the other side of the substrate. Another possibility for the inverted stereospecificity is that the substrate is bound in a different orientation. The crystal structure of D170S/T457E in complex with trifluoromethylphenol reveals that this possibility is rather unlikely. In the double mutant, the carboxylic moiety of Glu-457 is positioned at the opposite face of the substrate analog and only 3.5 Å from the Cα-atom. This indicates that, in the double mutants, Glu-457 directs the stereospecific water attack to the planar quinone methide intermediate, presumably by acting as an active site base (Fig. 5). In this respect, it is important to note that the single mutant T457E exhibits a strong preference for the formation of the (R)-enantiomer of 1-(4′-hydroxyphenyl)ethanol. This preference suggests that, in this particular mutant, Asp-170 favorably competes with Glu-457 for the site of water attack. The (R)-selectivity of T457E is in marked contrast with the stereochemical properties of PCMH. Like T457E, PCMH contains two acidic residues at opposite faces of the substrate binding site (4) but hydroxylates 4-ethylphenol preferably to (S)-1-(4′-hydroxyphenyl)ethanol (15, 16). Further mutagenesis studies are needed to establish whether the opposite stereospecificity of PCMH and T457E is related to the different type of acidic side chains (i.e., Glu-380 and Asp-170, respectively). Nevertheless, the above data show that subtle variations in active site topology determine the outcome of the enzyme stereospecificity and reinforce the idea that inversion of enantioselectivity can be generally carried out without major structural reconstruction (36). Our results are also in line with studies from medium-chain acyl-CoA dehydrogenase, where it was demonstrated that a functional glutamate involved in substrate dehydrogenation can be relocated in the enzyme active site by a double amino acid substitution (37, 38).
Figure 5.
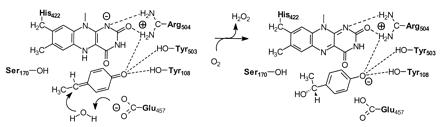
Schematic drawing of the hydration of the planar p-quinone methide intermediate of 4-ethylphenol in D170S/T457E. We propose that the Glu-457-activated water attacks the quinone methide, yielding the (S)-enantiomer of the alcohol.
The results presented here underline the important role of acidic residues for the function of VAO. In summary, Glu-457 can replace Asp-170 with respect to covalent flavin attachment, but not with respect to efficient redox catalysis. The stereospecificity of VAO depends on the position of the acidic residue in the enzyme active site. With Asp-170 present, VAO is highly stereospecific for the production of (R)-alcohols. However, the enzyme becomes (S)-selective when the acidic residue is relocated to the opposite face of the substrate-binding site.
Acknowledgments
This study was performed within the framework of the Innovation Oriented Research Program (IOP) Catalysis of the Dutch Ministry of Economy Affairs (Project IKA 96005). This work was supported under the Training and Mobility of Researchers/Access to Large-Scale Facilities program of the EMBL Hamburg Outstation, by Ministero dell' Universita e Ricerca Scientifica e Tecnologica (Project Biosintesi del nicotinamide dinucleotide: studi biochimici, biologia, strutturale e sviluppo razionale di farmaci), and Consiglio Nazionale delle Ricerche (CNR Target Project on Biotechnology).
Abbreviations
- PCMH
p-cresol methylhydroxylase
- VAO
vanillyl-alcohol oxidase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Macromolecular Structure Database of the European Bioinformatics Institute (EBI), Hinxton, United Kingdom (EBI ID code 1e0y).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160175897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160175897
References
- 1.Fraaije M W, van Berkel W J H, Benen J A E, Visser J, Mattevi A. Trends Biochem Sci. 1998;23:206–207. doi: 10.1016/s0968-0004(98)01210-9. [DOI] [PubMed] [Google Scholar]
- 2.Benson T E, Filman D J, Walsh C T, Hogle J M. Nat Struct Biol. 1995;2:644–653. doi: 10.1038/nsb0895-644. [DOI] [PubMed] [Google Scholar]
- 3.Dobbek H, Gremer L, Meyer O, Huber R. Proc Natl Acad Sci USA. 1999;96:8884–8889. doi: 10.1073/pnas.96.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunane L M, Chen Z-W, Shamala N, Mathews F S, Cronin C N, McIntire W S. J Mol Biol. 2000;295:357–374. doi: 10.1006/jmbi.1999.3290. [DOI] [PubMed] [Google Scholar]
- 5.Fraaije M W, Veeger C, van Berkel W J H. Eur J Biochem. 1995;234:271–277. doi: 10.1111/j.1432-1033.1995.271_c.x. [DOI] [PubMed] [Google Scholar]
- 6.van den Heuvel R H H, Fraaije M W, van Berkel W J H. J Bacteriol. 1998;180:5646–5651. doi: 10.1128/jb.180.21.5646-5651.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraaije M W, van Berkel W J H. J Biol Chem. 1997;272:18111–18116. doi: 10.1074/jbc.272.29.18111. [DOI] [PubMed] [Google Scholar]
- 8.Mattevi A, Fraaije M W, Mozzarelli A, Olivi L, Coda A, van Berkel W J H. Structure. 1997;5:907–920. doi: 10.1016/s0969-2126(97)00245-1. [DOI] [PubMed] [Google Scholar]
- 9.Mewies M, McIntire W S, Scrutton N S. Protein Sci. 1998;7:7–20. doi: 10.1002/pro.5560070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraaije M W, van den Heuvel R H H, van Berkel W J H, Mattevi A. J Biol Chem. 1999;274:35514–35520. doi: 10.1074/jbc.274.50.35514. [DOI] [PubMed] [Google Scholar]
- 11.van den Heuvel R H H, Fraaije M W, Mattevi A, van Berkel W J H. J Biol Chem. 2000;275:14799–14808. doi: 10.1074/jbc.275.20.14799. [DOI] [PubMed] [Google Scholar]
- 12.Trickey P, Wagner M A, Jorns M S, Mathews F S. Struct Fold Des. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 13.McIntire W S, Hopper D J, Singer T P. Biochem J. 1985;228:325–335. doi: 10.1042/bj2280325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drijfhout F P, Fraaije M W, Jongejan H, van Berkel W J H, Franssen M C R. Biotechnol Bioeng. 1998;59:171–177. doi: 10.1002/(sici)1097-0290(19980720)59:2<171::aid-bit5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 15.McIntire W S, Hopper D J, Craig J C, Everthart E T, Webster R V, Causer M J, Singer T P. Biochem J. 1984;224:617–621. doi: 10.1042/bj2240617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIntire W S, Bohmont C. In: Flavins and Flavoproteins. Edmondson D E, McCormick D B, editors. Berlin: Walter De Gruyter; 1987. pp. 677–686. [Google Scholar]
- 17.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 18.Woodcock D M, Crowther P J, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith S S, Michael M Z, Graham M W. Nucleic Acids Res. 1989;17:3469–3478. doi: 10.1093/nar/17.9.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson T G. Ph.D. thesis. Cambridge: Univ. of Cambridge; 1984. [Google Scholar]
- 20.Benen J A E, Sánchez-Torres P, Wagemaker M, Fraaije M W, van Berkel W J H, Visser J. J Biol Chem. 1998;273:7865–7872. doi: 10.1074/jbc.273.14.7865. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Fraaije M W, Pikkemaat M, van Berkel W J H. Appl Environ Microbiol. 1997;63:435–439. doi: 10.1128/aem.63.2.435-439.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massey V. In: Flavins and Flavoproteins. Curti B, Ronchi S, Zannetti G, editors. New York: Walter De Gruyter; 1991. pp. 59–66. [Google Scholar]
- 24.Abrahams J P, Leslie A G W. Acta Crystallogr Sec D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 25.Collaborative Computational Project Number 4. Acta Crystallogr Sec D. 1994;50:760–767. [Google Scholar]
- 26.Murshudov G N, Vagin A A, Dodson E J. Acta Crytallogr Sec D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Lamzin V S, Wilson K S. Acta Crystallogr Sec D. 1993;49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 28.Jones T A, Zou J Y, Cowan S W, Kjelgaard M. Acta Crystallogr Sec A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.de Jong E, van Berkel W J H, van der Zwan R P, de Bont J A M. Eur J Biochem. 1992;208:651–657. doi: 10.1111/j.1432-1033.1992.tb17231.x. [DOI] [PubMed] [Google Scholar]
- 30.Fraaije M W, Mattevi A, van Berkel W J H. FEBS Lett. 1997;402:33–35. doi: 10.1016/s0014-5793(96)01494-9. [DOI] [PubMed] [Google Scholar]
- 31.van Berkel W J H, van den Heuvel R H H, Versluis C, Heck A J R. Protein Sci. 2000;9:435–439. doi: 10.1110/ps.9.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark W M. Oxidation-Reduction Potentials of Organic Systems. Baltimore: Williams & Wilkins; 1960. pp. 184–203. [Google Scholar]
- 33.Minnaert K. Biochim Biophys Acta. 1965;110:42–56. doi: 10.1016/s0926-6593(65)80093-5. [DOI] [PubMed] [Google Scholar]
- 34.Frost J W, Rastetter W H. J Am Chem Soc. 1980;102:7157–7159. [Google Scholar]
- 35.Walsh C. In: Flavins and Flavoproteins. Massey V, Williams C H Jr, editors. Amsterdam: Elsevier Science; 1982. pp. 121–132. [Google Scholar]
- 36.May O, Nguyen P T, Arnold F H. Nat Biotechnol. 2000;18:317–320. doi: 10.1038/73773. [DOI] [PubMed] [Google Scholar]
- 37.Lee H-J K, Wang M, Paschke R, Nandy A, Ghisla S, Kim J-J P. Biochemistry. 1996;35:12412–12420. doi: 10.1021/bi9607867. [DOI] [PubMed] [Google Scholar]
- 38.Nandy A, Kieweg V, Kräutle F-G, Vock P, Küchler B, Bross P, Kim J-J P, Rasched I, Ghisla S. Biochemistry. 1996;35:12402–12411. doi: 10.1021/bi960785e. [DOI] [PubMed] [Google Scholar]