

Abstract
The high sensitivity of the magnetic resonance properties of xenon to its local chemical environment and the large 129Xe NMR signals attainable through optical pumping have motivated the use of xenon as a probe of macromolecular structure and dynamics. In the present work, we report evidence for nonspecific interactions between xenon and the exterior of myoglobin in aqueous solution, in addition to a previously reported internal binding interaction. 129Xe chemical shift measurements in denatured myoglobin solutions and under native conditions with varying xenon concentrations confirm the presence of nonspecific interactions. Titration data are modeled quantitatively with treatment of the nonspecific interactions as weak binding sites. Using laser-polarized xenon to measure 129Xe spin-lattice relaxation times (T1), we observed a shorter T1 in the presence of 1 mM denatured apomyoglobin in 6 M deuterated urea (T1 = 59 ± 1 s) compared with that in 6 M deuterated urea alone (T1 = 291 ± 2 s), suggesting that nonspecific xenon-protein interactions can enhance 129Xe relaxation. An even shorter T1 was measured in 1 mM apomyoglobin in D2O (T1 = 15 ± 0.3 s), compared with that in D2O alone (T1 = 506 ± 5 s). This difference in relaxation efficiency likely results from couplings between laser-polarized xenon and protons in the binding cavity of apomyoglobin that may permit the transfer of polarization between these nuclei via the nuclear Overhauser effect.
The magnetic resonance properties of xenon demonstrate remarkable sensitivity to its local chemical environment, making the noble gas an effective NMR probe of molecules, materials, and biological systems (1–4). The hydrophobicity of xenon makes it particularly well suited for exploring the nonpolar interiors of macromolecules, proteins, and membranes that bind xenon in solution. Several studies have exploited sensitive xenon chemical shifts to characterize the interactions between xenon and organic compounds such as α-cyclodextrin (5) and cryptophane-A (6), the membrane protein channel gramicidin (7), and the globular proteins myoglobin and hemoglobin (8). In addition, it has been shown that the 129Xe spin-lattice relaxation time (T1) is reduced dramatically when xenon comes into contact with cyclodextrins (9), lipids (10), and proteins in blood (11–15). Recent advances in optical pumping technology (16) have further motivated such studies of xenon-molecule interactions by providing 129Xe NMR signal enhancements of up to five orders of magnitude (17). Furthermore, the transfer of spin polarization from laser-polarized xenon to nearby nuclei through the spin polarization induced nuclear Overhauser effect (18) allows direct identification of xenon binding sites and reveals information concerning the structure and dynamics of xenon-molecule complexes (19–21).
Hydrophobic regions of proteins play a fundamental role in the formation of protein-protein and protein-lipid interfaces and in the stability of many small ligands within protein binding sites. X-ray crystallography studies of protein-xenon complexes have demonstrated the accessibility of protein interiors to xenon (22–25). Accordingly, characterization of the interactions of xenon with proteins is a promising approach to studying biomolecular interfaces and ligand-protein binding. The crystal structure of myoglobin in the presence of 7 atm (1 atm= 101.3 kPa) of xenon gas exhibits four xenon binding sites, ranging from approximately half to full occupancy (24). The binding constants of the two major sites have been determined by manometry to be 190 M−1 and 10 M−1, with the strongest site most likely corresponding to the cavity in the crystal structure near the heme (24, 26).
NMR studies of the chemical shift, relaxation, and polarization transfer properties of xenon in protein solutions are capable of providing information toward the mapping of hydrophobic surfaces in proteins and the modeling of their interactions with ligands. Tilton and Kuntz first made chemical shift measurements of xenon in metmyoglobin solutions, observing a single 129Xe resonance (at room temperature and 1 atm of Xe overpressure) caused by fast exchange of xenon between the solvent and the internal sites in the protein (8). The upfield changes in the shift with increasing metmyoglobin concentration were attributed to the contribution of the specifically bound xenon species to the overall shift. The 129Xe T1 in 0.1 mM metmyoglobin in D2O has been measured to be 1.3 s at 4.7 T, much shorter than the T1 of 470 s observed at the same field in D2O alone (27). It was concluded that the short T1 arises from efficient relaxation pathways available to xenon bound in the protein. Competitive ligand studies have been used to demonstrate that a significant source of relaxation of laser-polarized xenon in blood plasma is the binding of xenon to albumin proteins (14). 129Xe NMR has also been used to probe the interactions between adsorbed laser-polarized xenon and the surfaces of lyophilized protein solids (28). In the present work, we report evidence of nonspecific interactions between xenon and the exterior of myoglobin in solution that affect 129Xe chemical shifts and spin-lattice relaxation times, in addition to the previously reported internal binding interactions. Such nonspecific interactions between proteins and small molecule volatile anesthetics, of which xenon is a known example (29, 30), may play a role in the biochemistry of anesthesia (31, 32). Characterization of these nonspecific effects at protein surfaces could permit the use of xenon as a molecular probe of protein-solvent and protein-protein interfaces.
Materials and Methods
Protein samples used in titration studies were prepared by dissolving lyophilized horse skeletal muscle myoglobin powder (Sigma) into water that contained 20% D2O. Concentrations were determined by absorption of 280 nm UV light using ɛmyoglobin = 31,000 M−1⋅cm−1 and ɛapomyoglobin = 13,980 M−1⋅cm−1 (33, 34). Apomyoglobin was prepared by reverse phase HPLC. The apoprotein and heme were eluted as separate fractions after application of the holoprotein to the column. Solvent was evaporated from the protein fraction, and the remaining powder was redissolved and dialyzed against water before lyophilization. The absence of the heme was confirmed by the disappearance of its characteristic absorption band at ≈409 nm. Relaxation measurements were made in solutions of apomyoglobin in 99.96% D2O with urea-D4 as a protein denaturant.
Chemical shift measurements were made using 80% enriched 129Xe (Isotec) that was loaded into medium wall NMR tubes (Wilmad, Buena, NY) adapted for the moderately high pressures used in these experiments; pressures were confirmed with a gauge upon loading and subsequent recovery of the gas after each experiment. Samples were gently agitated and allowed to equilibrate for approximately 1 h before acquisition of data with a Bruker AMX 600 MHz spectrometer at 25°C. Xenon concentrations in water ([Xe]o) were assumed to be linear with gas overpressure with a slope of 4.37 mM/atm (35). A sealed glass capillary tube filled with ≈7 atm of xenon gas was included in each sample as a standard reference to ensure that changes in 129Xe chemical shifts were not caused by artifacts induced during the acquisition or processing of titration data. 129Xe spectra were fit to Lorentzian lineshapes with the felix 97 NMR data processing software package (Molecular Simulations, Waltham, MA) to quantify chemical shifts more accurately. Each data set was processed and fit at least five times, and the standard deviation of the shift is reported as the error estimate of the chemical shift.
Spin lattice relaxation times were measured with optically pumped natural abundance xenon. After polarization, the xenon was transferred into the sample tube, and the solution was shaken to achieve rapid equilibration. Xenon polarizations were typically ≈10%. Xenon signal intensities were measured by using small pulses (3° tipping angle) that were calibrated by using a pressurized xenon-zeolite sample. Spectra were acquired by using a Varian INOVA 300 MHz spectrometer, and integrated peak intensities were fit to an exponential decay to determine relaxation times. The reported 129Xe T1s are corrected for the loss of z-magnetization from the small RF pulses. Samples used in relaxation experiments were thoroughly degassed, and the glass tubes were treated with a siliconizing agent.
Results and Discussion
The chemical shift of xenon in metmyoglobin solutions of different concentration was measured at a xenon pressure of 5 atm (Figs. 1 and 2A). At this relatively high xenon concentration in solution, we observed that the shift moves downfield from water with increasing protein concentration but that it seems to have an upfield component that becomes significant at higher concentrations. The behavior of the observed shift with increasing protein concentration suggests multiple interactions between xenon and the protein with opposing effects on the overall shift. However, the fraction of xenon occupying the second binding site (K = 10 M−1) (26) is calculated to be small at the concentrations of xenon and protein used in the titration. Accordingly, we propose that the downfield contribution to the observed shift arises from nonspecific interactions between xenon and the exterior of the protein whereas the upfield contribution comes from xenon bound to the previously reported specific site. Evidence for xenon-protein interactions at protein surfaces has been obtained through x-ray crystallography (25) and 129Xe NMR of lyophilized protein solids (28). In addition, weak nonspecific interactions have been reported for xenon-cyclodextrin solutions, and the effect on the xenon chemical shift was assumed to be linear in cyclodextrin concentration (5).
Figure 1.
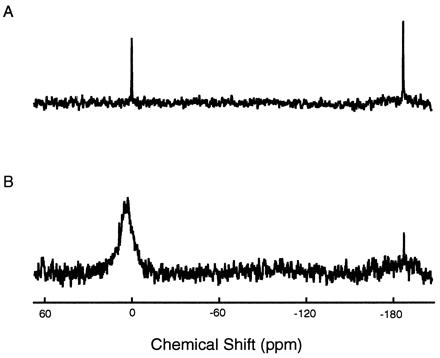
Typical 129Xe NMR spectra of thermally polarized xenon gas dissolved in solvent (80% H2O/20% D2O) (A) and a 10 mM metmyoglobin solution under ≈5 atm of xenon overpressure (B). Chemical shifts are referenced to the shift of 129Xe in solvent. The upfield peak corresponds to xenon gas filling a capillary tube placed within the samples for in situ referencing.
Figure 2.
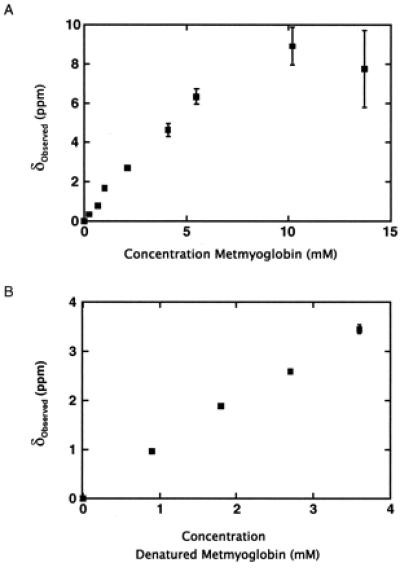
(A) Xenon chemical shifts in metmyoglobin solutions under ≈5 atm of xenon overpressure referenced to the shift in 80% H2O/20% D2O. Error bars correspond to fitting errors (see Materials and Methods). (B) Xenon chemical shifts in denatured myoglobin solutions under ≈5 atm of xenon overpressure referenced to the shift in 80% H2O/20% D2O and 6 M urea.
To confirm the presence of nonspecific xenon-protein interactions, xenon chemical shifts were also measured as a function of denatured protein concentration (Fig. 2B). The unfolded protein in 6 M urea does not contain the specific xenon binding sites present in the native structure but can still interact nonspecifically with the dissolved gas. Indeed, the observed xenon chemical shift, referenced to its shift in solvent, moves downfield with increasing denatured protein concentration. A comparison of the effect of nonspecific interactions under native versus denaturing conditions can be made by considering the initial slope of both titrations; the initial slope of the native titration is the limit at which only a small fraction of xenon is bound to the specific site, and thus the overall shift is dominated by nonspecific interactions. As seen in Fig. 2, the slope of the native titration is steeper than the slope of the denatured titration, indicating that nonspecific interactions under native conditions make a different contribution to the chemical shift. This result is reasonable given that parts of the protein (e.g., hydrophobic side chains and the heme) that are previously inaccessible to solvent become accessible to nonspecific interactions when the protein is unfolded. The fact that the initial slope has a smaller downfield magnitude in the denatured protein titration (despite an increased number of possible nonspecific interactions) suggests that nonspecific interactions between xenon and the hydrophobic residues and heme within the previously hidden interior of the protein induce a xenon chemical shift that is upfield relative to the chemical shift induced by interactions between xenon and surface residues. The detection of weak nonspecific interactions with different effects on the xenon chemical shift in these titrations is made possible by the high sensitivity of the shift to the local environment of xenon and illustrates the ability of xenon to probe diverse molecular interactions.
The downfield shifts in the native metmyoglobin titration at high xenon concentration are in contrast to the upfield shifts previously reported by Tilton and Kuntz for samples with low xenon concentration (8). The differences in data collected under varying xenon concentrations demonstrate the opposing effects of nonspecific and specific interactions on the overall 129Xe chemical shift. At low xenon concentration, the specific binding contribution to the average chemical shift dominates the contribution from the small amount of free xenon interacting nonspecifically so that the 129Xe shift is always upfield from its shift in aqueous solvent. Conversely, at high xenon concentrations, the nonspecific interactions between the abundant free xenon and the numerous protein surface sites result in an overall downfield shift from solvent. We obtained experimental evidence for the marked xenon concentration dependence of the chemical shift resulting from nonspecific interactions using a 5 mM metmyoglobin sample (Fig. 3). Although the 129Xe shift is upfield (≈−6 ppm) at low xenon concentrations (≈4 mM), the xenon chemical shift was observed to move downfield with increasing total xenon concentration as the internal binding site becomes saturated, and additional free xenon—capable of interacting nonspecifically with the protein surface—is introduced into solution. Nonspecific interactions result in a limiting chemical shift at high xenon concentration that is further downfield than the shift of xenon in aqueous solvent; if only specific binding were present, one would expect this limiting shift to be the 129Xe shift in solvent.
Figure 3.
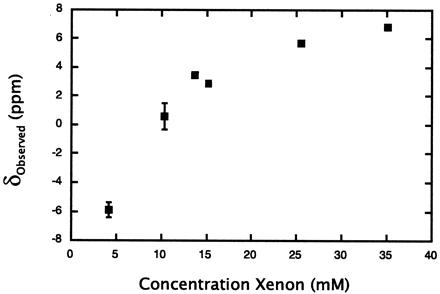
The effect of total xenon concentration on 129Xe chemical shift titrations. Experimental chemical shifts, referenced to the shift of 129Xe in solvent (80% H2O/20% D2O), obtained with varying xenon concentrations at a metmyoglobin concentration of 5 mM. The concentration of xenon is assumed to depend linearly on the gas overpressure with a slope of 4.37 ppm/atm (35).
A simple model was constructed to use the native protein titration data to quantify the strengths of both specific and nonspecific interactions and their effects on the observed xenon chemical shift. The observed shift is a weighted average of the chemical shift of xenon in solvent, xenon bound specifically within the interior of the native protein, and nonspecific xenon-protein interactions that can be treated as weak binding sites. Accordingly, assuming the limit at which a small fraction of xenon is bound to the nonspecific sites, the expression for the observed xenon chemical shift referenced to the shift in solvent can be written as
 |
1 |
where [Xe]bound depends on the strength of binding to the specific site (Kbound), and
 |
2 |
Thus, α represents the total contribution of the nonspecific interactions to the observed chemical shift.
To calculate the unknown parameters in Eq. 1, an assumption must be made concerning the total xenon concentration in solution. In one limit, the total xenon concentration can be considered to be independent of protein concentration and equal to the concentration of xenon in solvent ([Xe]total = [Xe]o). Fitting the data in Fig. 2 to this model in which [Xe]o = 23 mM yields a value of δbound = −40 ppm, Kbound = 140 M−1, and α = 3 ppm/mM for the native protein titration and α = 1 ppm/mM for the titration under denaturing conditions. The values of 140 M−1 for Kbound and −40 ppm for δbound are in close agreement with those previously reported for the strongest specific site [190 M−1 (26) and −43 ppm (8), respectively]. In the other limit, the total xenon concentration in solution is assumed to be the sum of the concentration of xenon in solvent plus the concentration of xenon bound to the specific site ([Xe]total = [Xe]o + [Xe]bound). Using this model, we have found that the data can only be fit uniquely if the specific binding constant (Kbound) is fixed to the value previously reported (26); accordingly, the fit yields a value of δbound = −20 ppm and α = 2 ppm/mM. Although Eq. 1 considers a protein with a single specific binding site, appropriate for myoglobin because the other sites observed in the crystal structure have low occupancies even at relatively high xenon pressures (24), the models described here can be generalized to proteins and other molecules with multiple specific binding sites.
We have used laser-polarized xenon to measure 129Xe T1s in the presence of apomyoglobin under both native and denaturing conditions to examine the effect of both specific internal binding and nonspecific surface interactions on xenon spin-lattice relaxation. We have measured the decay of xenon signal in several solutions as a function of time (Fig. 4). The strong NMR signals (Fig. 4 Inset) are attributable to the high xenon polarization obtained with optical pumping and allow precise determination of relaxation times. The observed 129Xe T1 was only 15.0 ± 0.3 s in 1 mM apomyoglobin in D2O compared with 506 ± 5 s measured in D2O alone. This confirms the presence of interactions between xenon and the protein that provide efficient mechanisms for relaxation. The T1 in 1 mM apomyoglobin is still an order of magnitude longer than that measured by Bryant and coworkers in only a 0.1 mM solution of metmyoglobin (27). This variance in relaxation may partially arise from differences in the binding affinities of the holoprotein and apoprotein for xenon (currently under investigation in our laboratory). However, a more dominant cause of the shorter xenon T1 in metmyoglobin solutions is likely the strong interaction of the bound xenon and paramagnetic iron within the nearby heme.
Figure 4.
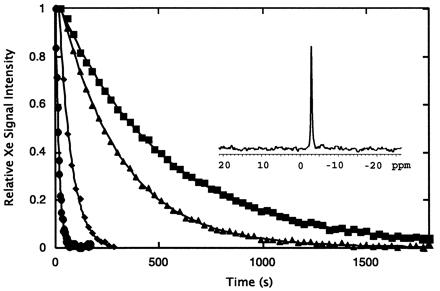
Integrated intensities of laser-polarized 129Xe NMR signals as a function of time for xenon dissolved in D2O (■), with 1 mM apomyoglobin in D2O (●), with 6 M deuterated urea in D2O (▴), and with 1 mM apomyoglobin and 6 M urea-D4 in D2O (♦). Calculated spin-lattice relaxation times for 129Xe in these solutions are 506 ± 5 s, 15.0 ± 0.3 s, 291 ± 2 s, and 59 ± 1 s, respectively. The inset shows the typical signal-to-noise with a single 3° pulse after the dissolution of laser-polarized xenon into a 1 mM solution of apomyoglobin; the shift is referenced to that in pure D2O.
To determine the effect of the nonspecific interactions detected in chemical shift experiments on xenon relaxation times, we measured the T1 of laser-polarized 129Xe in a solution of apomyoglobin and 6 M deuterated urea. Compared with 291 ± 2 s in 6 M deuterated urea alone, a shorter relaxation time of 59 ± 1 s in the presence of 1 mM denatured apomyoglobin demonstrates that nonspecific xenon-protein interactions contribute to xenon spin-lattice relaxation. However, the T1 in 1 mM apomyoglobin under native conditions is still substantially shorter than under denaturing conditions, despite the presence of an increased number of potential nonspecific interactions upon unfolding and the presence of urea in the denatured case. This difference in relaxation efficiency likely results from significant couplings between specifically bound xenon and protons in the binding cavity within the native structure. The cross relaxation indirectly observed in these experiments may permit the observation of polarization transfer via the nuclear Overhauser effect from laser-polarized xenon to protons in apomyoglobin. Work is in progress to effect this polarization transfer, with the ultimate goal of mapping hydrophobic surfaces in proteins and characterizing the structure and dynamics of ligand-protein interactions.
Acknowledgments
The authors thank Tom Lawhead for his expert glassblowing and advice. S.M.R. and M.M.S. acknowledge the National Science Foundation for predoctoral fellowships. This work was supported by the Director, Office of Energy Research, Office of Basic Energy Sciences, Materials Sciences Division, Physical Biosciences Division, of the U.S. Department of Energy under Contract No. DE-AC03-765F00098.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170278897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170278897
References
- 1.Miller K W, Reo N V, Schoot-Uiterkamp A J M, Stengle D P, Stengle T R, Williamson K L. Proc Natl Acad Sci USA. 1981;78:4946–4949. doi: 10.1073/pnas.78.8.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraissard J, Ito T. Zeolites. 1988;8:350–361. [Google Scholar]
- 3.Jokisaari J. Prog Nucl Magn Reson Spectrosc. 1994;26:1–26. [Google Scholar]
- 4.Ratcliffe C. Annu Rep NMR Spectrosc. 1998;36:124–208. [Google Scholar]
- 5.Bartik K, Luhmer M, Heyes S J, Ottinger R, Reisse J. J Magn Reson Ser B. 1995;109:164–168. [Google Scholar]
- 6.Bartik K, Luhmer M, Dutasta J P, Collet A, Reisse J. J Am Chem Soc. 1998;120:784–791. [Google Scholar]
- 7.McKim S, Hinton J F. Biochim Biophys Acta. 1994;1193:186–198. doi: 10.1016/0005-2736(94)90348-4. [DOI] [PubMed] [Google Scholar]
- 8.Tilton R F, Jr, Kuntz I D., Jr Biochemistry. 1982;21:6850–6857. doi: 10.1021/bi00269a035. [DOI] [PubMed] [Google Scholar]
- 9.Hitchens T K, Bryant R G. J Magn Reson. 1997;124:227. [Google Scholar]
- 10.Xu Y, Tang P. Biochim Biophys Acta. 1997;1323:154–162. doi: 10.1016/s0005-2736(96)00184-8. [DOI] [PubMed] [Google Scholar]
- 11.Albert M S, Schepkin V D, Budinger T F. J Comput Assist Tomogr. 1995;19:975–978. doi: 10.1097/00004728-199511000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Bifone A, Song Y-Q, Seydoux R, Taylor R E, Goodson B M, Pietrass T, Budinger T F, Navon G, Pines A. Proc Natl Acad Sci USA. 1996;93:12932–12936. doi: 10.1073/pnas.93.23.12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tseng C H, Peled S, Nascimben L, Oteiza E, Walsworth R L, Jolesz F A. J Magn Reson. 1997;126:79–86. doi: 10.1006/jmre.1997.1145. [DOI] [PubMed] [Google Scholar]
- 14.Wolber J, Cherubini A, Dzik-Juraz A S K, Leach M O, Bifone A. Proc Natl Acad Sci USA. 1999;96:3664–3669. doi: 10.1073/pnas.96.7.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albert M S, Kacher D F, Balamore D, Venkatesh A K, Jolesz F A. J Magn Reson. 1999;140:264–273. doi: 10.1006/jmre.1999.1836. [DOI] [PubMed] [Google Scholar]
- 16.Walker T G, Happer W. Rev Mod Phys. 1997;69:629–642. [Google Scholar]
- 17.Goodson B M, Kaiser L, Pines A. Proc Int School Phys “Enrico Fermi”. 1999;139:211–260. [Google Scholar]
- 18.Navon G, Song Y-Q, Rõõm T, Appelt S, Taylor R E, Pines A. Science. 1996;271:1848–1851. [Google Scholar]
- 19.Song Y-Q, Goodson B M, Taylor R E, Laws D D, Navon G, Pines A. Angew Chem Int Ed Engl. 1997;36:2368–2370. [Google Scholar]
- 20.Luhmer M, Goodson B M, Song Y-Q, Laws D D, Kaiser L, Cyrier M C, Pines A. J Am Chem Soc. 1999;121:3502–3512. [Google Scholar]
- 21.Song Y-Q. Concepts Magn Reson. 2000;12:6–20. [Google Scholar]
- 22.Schoenborn B P, Watson H C, Kendrew J C. Nature (London) 1965;207:28–30. doi: 10.1038/207028a0. [DOI] [PubMed] [Google Scholar]
- 23.Schoenborn B P. Nature (London) 1965;208:760–762. doi: 10.1038/208760a0. [DOI] [PubMed] [Google Scholar]
- 24.Tilton R F, Jr, Kuntz I D, Jr, Petsko G A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 25.Prangé T, Shiltz M, Pernot L, Colloc'h N, Longhi S, Bourget W, Fourme R. Struct Funct Genet. 1998;30:61–73. [PubMed] [Google Scholar]
- 26.Ewing G J, Maestas S. J Phys Chem. 1970;74:2341–2344. doi: 10.1021/j100705a018. [DOI] [PubMed] [Google Scholar]
- 27.Stith A, Hitchens T K, Hinton D P, Berr S S, Driehuys B, Brookeman J R, Bryant R G. J Magn Reson. 1999;139:225–231. doi: 10.1006/jmre.1999.1781. [DOI] [PubMed] [Google Scholar]
- 28.Bowers C R, Storhaug V, Webster C E, Bharatam J, Cottone A, III, Gianna R, Betsey K, Gaffney B J. J Am Chem Soc. 1999;121:9370–9377. doi: 10.1021/ja991443+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullen S C, Gross E G. Science. 1951;113:580–582. doi: 10.1126/science.113.2942.580. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy R R, Stokes J W, Downing P. Anaesth Intensive Care. 1992;20:66–70. doi: 10.1177/0310057X9202000113. [DOI] [PubMed] [Google Scholar]
- 31.Eckenhoff R G, Johansson J S. Pharmacol Rev. 1997;49:343–367. [PubMed] [Google Scholar]
- 32.Eckenhoff R G, Tanner J W. Biophys J. 1998;75:477–483. doi: 10.1016/S0006-3495(98)77536-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breslow E. J Biol Chem. 1964;239:486–496. [PubMed] [Google Scholar]
- 34.Pace C N, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clever H L, editor. Solubility Data Series. New York: Pergamon; 1979. [Google Scholar]