

Abstract
The crystal structure of a B-type DNA hexanucleotide duplex complexed with the porphyrin molecule nickel-[tetra-N-methyl-pyridyl] porphyrin has been solved by multiwavelength anomalous diffraction phasing and refined to an R factor of 11.5% at a resolution of 0.9 Å. The structure has been solved and refined as two crystallographically independent duplexes, stacked end to end. Contrary to expectation, the porphyrin molecule is not intercalated into the duplex but is stacked onto the ends of the two-duplex stack. The porphyrin molecule is highly buckled as a consequence of the nickel coordination, which produces large changes in local DNA structure. A second mode of porphyrin binding is apparent as a consequence of crystal packing, which places the ligand in the minor groove of an adjacent duplex. This structure thus provides, to our knowledge, the first atomic visualization of minor-groove binding for a porphyrin molecule. The geometry of groove binding provides a ready explanation for porphyrin-induced DNA strand cleavage at deoxyribose residues.
Keywords: DNA structure, minor groove binding, end capping
The interactions of cationic porphyrins with nucleic acids have received considerable attention (1, 2). Several have clinical potential as anticancer agents in photodynamic therapy (3, 4), probably as a consequence of their ability to selectively accumulate on the surface of tumor cells, become internalized, bind to genomic DNA, and then induce DNA strand cleavage. One such compound is tetra-(N-methylpyridyl) porphyrin (TMPy: Fig. 1a), which can be coordinated with a range of transition metals (1, 2, 5, 6). TMPy has more direct antitumor activity, possibly as a consequence of its ability to inhibit the telomerase enzyme (7). Binding to quadruplex DNA as well as to duplexes has been reported for TMPy and various complexes (8–10). Stabilization of G-quadruplex structures has been shown to be the mechanism whereby TMPy inhibits telomerase from catalyzing the synthesis of further linear telomere repeats. NMR and photocleavage data have been interpreted in favor of a model with a TMPy molecule externally stacking onto guanine–quadruplex structures (8–10).
Figure 1.
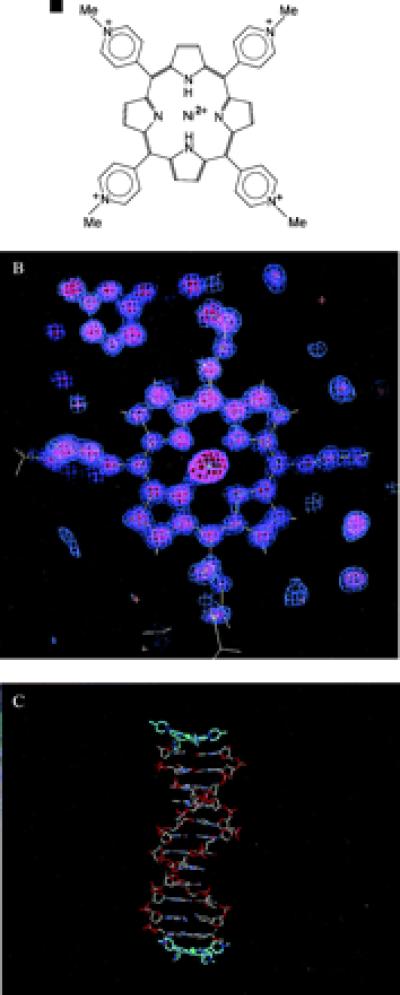
(a) The structure of the Ni2+-TMPy ligand. (b) Final 2Fo − Fc electron density map, calculated in the plane of the Ni2+-TMPy ligand. (c) plot of the 2:2 complex in the crystallographic asymmetric unit.
Noncovalent interactions of TMPy and its metal complexes with duplex DNA are sequence dependent (2, 11), with intercalation occurring at 5′-CpG sites, as shown by NMR and other biophysical studies (12, 13) as well as molecular modeling (13, 14). A crystallographic analysis (15) of the Cu2+ complex of TMPy bound to the sequence 5′-d(CGTACG)2 also found the ligand intercalated at this site, but with the 5′-end cytosine swung out in an extrahelical arrangement. By contrast, biophysical and modeling (14, 16) studies have suggested that TMPy itself (and many of its metal complexes) bind in the minor groove at A/T sequences and perturb the double helix only to a minimal extent at such sites (16). No structural data has been reported to date on groove-binding modes. TMPy itself is a planar molecule (14, 15), and it has been assumed that the groove binding would therefore be analogous to other planar small-molecule ligands.
We report here the crystallographic analysis of Ni2+-TMPy bound to the sequence 5′-d(CCTAGG). The structure of the TMPy-d(CCTAGG) complex is very well ordered, with a densely packed crystal lattice. This arrangement has resulted in exceptionally highly diffracting crystals and has enabled us to refine the structure to a resolution of 0.9 Å. The final stage of refinement was performed by using full-matrix methods, which have yielded meaningful estimated standard deviations for atomic coordinates and hence for derived geometric parameters.
Materials and Methods
Crystallization.
The sequence 5′-d(BrCCTAGG) was obtained from Oswel DNA Service (University of Southampton, Southampton, United Kingdom). Nickel2+-meso-tetra [4-N-methyl(pyridyl)] porphyrin (Ni2+-TMPy) was provided by David Wilson, Department of Chemistry, Georgia State University, Atlanta, GA. Large crystals were grown by vapor diffusion from a 12-μl drop containing 1.25 mM DNA, 1.25 mM Ni2+-TMPy, 50 mM MgCl2, 5% MPD, and 30 mM sodium cacodylate at pH 7.0, equilibrated against a 500-μl solution of 55% MPD.
Data Collection.
Fluoresence measurements and multiwavelength anomalous diffraction (MAD) data were collected at the European Synchroton Radiation Facility, Station BM14, Grenoble, France. High-resolution data were collected at Station X11 of the Deutsches Elektronen Synchrotron, Hamburg, Germany. Before both data collections, crystals were flash frozen at 100 K, tested in-house for diffraction quality, and then transferred to and retrieved from storage by using a King's Cryogun, a portable cryostream that we have developed in the King's College laboratory.
Suitable wavelengths for the MAD experiment were chosen from a crystal fluorescence spectrum to be λ1 = 0.9189 Å (midpoint), λ2 = 0.9198 Å (maxima), and λ3 = 0.7747 Å (remote point). The fluorescence spectrum also indicated anisotropy of the anomalous signal and bromine atoms located in two different environments. The high-resolution data were collected by three passes of the crystal between the resolution ranges 0.9–1.8 Å, 1.6–3.0 Å, and 2.8–32 Å. Both the MAD and high-resolution data sets were collected on MAR345 detectors and processed with programs from the hkl suite (17), and all overloaded reflections were rejected. Merging intensity statistics for both data sets are shown in Table 1.
Table 1.
Crystallographic data
| MAD data | High-resolution data | |||
|---|---|---|---|---|
| Space group | P41 | |||
| Contents of asymmetric unit | 2 duplexes of d(CCTAGG) + 2 Ni-TMPy | |||
| Unit cell dimensions | a = 32.320 Å c = 62.157 Å | a = 32.210 Å c = 62.259 Å | ||
| Maximum resolution, Å | 1.59 | 1.59 | 1.19 | 0.86 |
| Wavelength, Å | 0.9198 | 0.9189 | 0.7747 | 0.9057 |
| No. of unique reflns* | 30,852 | 8,562 | 74,396 | 45,197 |
| |F(hkI)|>4σ | ||||
| Total no. of reflns | 46,441 | 44,830 | 120,055 | 336,951 |
| †Rmerge | 1.7% | 1.7% | 1.4% | 2.1% |
| ‡Ranom | 7.0% | 3.2% | 4.2% | 4.4% |
| Phasing statistics | (treated as native) | |||
| Phasing power, acentric/centric | 4.49/5.45 | 2.88/2.49 | ||
| RCullis, acentric/centric | 0.29/0.19 | 0.40/0.36 | ||
| Mean figure of merit, acentric/centric | 0.87/0.85 | |||
| Refinement statistics | ||||
| Refinement range, Å | 6.0–0.90 | |||
| R factor all reflns | 15.2% | |||
| No. of reflns | 45,197 | |||
| R factor for reflns >4σ(F) | 14.2% | |||
| No. of reflns | 37,075 | |||
| % completeness, R factor for highest-resolution shell | 70.3%, 17.4% | |||
| Free R factor | 16.1% | |||
| Total no. of least-squares parameters | 3,867 | |||
| Total no. of atoms | 2,360 | |||
| Number of water molecules | 198 | |||
| Number of magnesium ions | 2 | |||
reflns, reflections.
*Friedel pairs were kept separate for the MAD data and were merged for the high-resolution data used for the refinement.
Rmerge is defined as Σ|I − <I>|/ΣI with Bijvoet pairs being treated separately.
Ranom is defined as Rmerge with only Bijvoet pairs now being merged in the calculation.
Crystal Structure Determination.
Software from the ccp4 package (18) was used for solution of the structure. Analysis of anomalous, dispersive, and isomorphous difference Patterson maps revealed four unique bromine atoms in the asymmetric unit cell. Treating the MAD data at three wavelengths as multiple isomorphous replacement data with λ2 as native, the positional and temperature factors for the bromine atoms were refined and experimental phases and their figure of merit were calculated to 1.6 Å. The xtalview program suite (19) was used for building the structure to the electron density map calculated with these phases. This map was of high quality, with individual atomic centers clearly defined.
Model Refinement.
The program shelx97–2 (20) was used for the refinement of the structure. Standard bond lengths and bond angles for the DNA were taken from the Nucleic Acid Database (21). Those for the porphyrin were obtained from molecular mechanics calculations by using the MM2 force field (22). The starting model was put through 20 cycles of positional and isotropic temperature factor refinement by using distance and chiral volume restraints. σA-weighted Fo − Fc difference maps revealed two unique Mg2+ cations, each with hexacoordinated water molecules. A disordered region of phosphate backbone, one in each hexamer duplex, was identified and refined as having two distinct conformations, with sum occupancies equal to unity. The revised model was refined for a further 20 cycles. Peak picking in difference Fourier maps found a total of 198 acceptable potential water/ion peaks. Temperature factors, hydrogen-bonding distances, and values of Rfree were used to discriminate between spurious density and true solvent molecules. These were included in the model as water molecules and refined for 20 more cycles. Once all solute and solvent structure had been accounted for, the structure was refined for a further 20 cycles with anisotropic temperature factors for the P, Ni, and Mg atoms of the DNA, porphyrin molecules, and magnesium ions. Contributions from hydrogen atoms were included in the structure at this point by using a riding model, with 10 further cycles of refinement. Further refinement was then performed with all phosphate groups and water of hydration around the magnesium ions assigned anisotropic temperature factors.
At this stage, the restraints were removed, and full-matrix least-squares refinement was attempted on the complete structure. Detailed examination of the structure showed that this had not resulted in significant geometric perturbations from ideality, and that the values for R and Rfree had significantly improved. Statistics for the final refined structure are given in Table 1. Estimated standard deviations (esds) in geometric parameters were calculated directly from the least-squares covariance matrix. Esds in bond lengths are in the range 0.02–0.04 Å and in bond and torsion angles 1–4°. Positional and thermal parameters and structure factors have been deposited in the Protein Data Bank (www.rcsb.org), (PDB ID code 1EMO) and in the Nucleic Acid Database as accession code DD0027.
Results and Discussion
The Overall Structure.
The asymmetric unit of the crystal structure contains two duplexes of the self-complementary hexamer 5′-d(BrCCTAGG). These are packed together end to end in the crystal, forming an antiparallel pseudododecamer, with noncrystallographic 2-fold rotation symmetry. A Ni2+-TMPy molecule (Fig. 1b) is stacked onto each end of the pseudododecanucleotide, giving a 2:2 stoichiometry for the complex (Fig. 1c). The crystal structure has no further stacking of DNA molecules onto these ends. We initially expected the structure to be related to the intercalated type of arrangement in the 5′-d(CGTACG)-Cu2+-TMPy complex (15), especially because we presumed that minor-groove binding at A/T sequences would require three to four contiguous A:T base pairs (16). Instead, we find that the present complex has a distinctive nonintercalative stacked arrangement for the bound TMPy molecule (explaining why numerous attempts at structure solution by using intercalated models did not produce correct crystal structures).
The crystal packing arrangement for this structure also inserts these TMPy end-cap molecules into the minor grooves of symmetry-related duplexes (Figs. 2 a and b) and thus provides the first x-ray structural view on the minor-groove mode of binding for porphyrins. The minor-groove interactions of each TMPy molecule exclusively involve nonbonded contacts, with two N-methyl-pyridyl substituents buried deep into the groove. The porphyrin-binding site on one hexanucleotide duplex, of ≈3.5 base pair, is not symmetric. It completely covers the two A:T base pair as well as the 3′ adjacent C:G base pair. The groove-binding arrangement confirms that envisaged previously in outline by molecular modeling (16), although it has previously been assumed that the presence of the exocyclic N2 atom of guanine in the minor groove would hinder groove binding at C/G-containing sequences. The two modes observed in this structure, external stacking and groove binding, both have significant effects on local and global DNA structure, which are detailed below.
Figure 2.
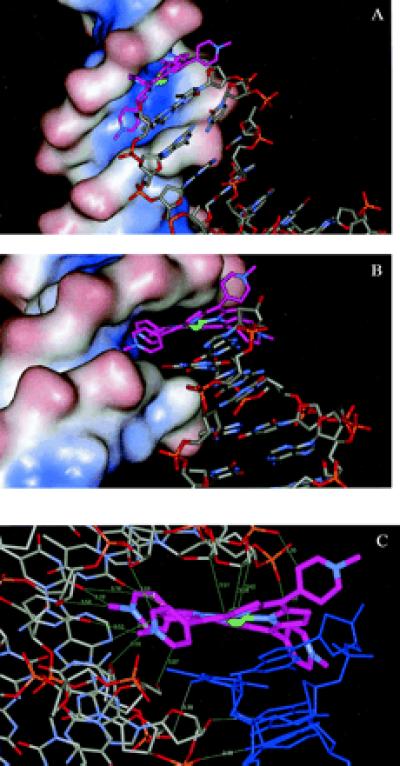
(a) A view of the minor-groove solvent-accessible surface of one duplex, together with a symmetry-related duplex whose terminal Ni2+-TMPy ligand is embedded into the minor groove of the first duplex. (b) A second view, now looking directly down the groove. The nickel atom is shown as a green sphere, and the carbon atoms of the TMPy molecule are shown in purple. (c) Detailed view of the nonbonded distances <3.5 Å (shown in green) between the Ni2+-TMPy molecule and the minor groove. The close distances between the minor groove and the “carrier” duplex (colored blue) are also shown in cyan.
In addition to the DNA–porphyrin complex, the two magnesium–water cations and the surrounding solvent also exhibit approximate noncrystallographic 2-fold symmetry. The solvent structure is extensive and shows considerable ordered structure. Noteworthy is the presence of an extended network of water molecules at the four C:G base pair of the minor-groove dimer interface (Fig. 3a), which is arranged approximately symmetrically around the water molecule W511 on the noncrystallographic 2-fold. A complete set of first- and second-shell water molecules and their interactions is seen, which links the two strands of the pseudoduplex. The network starts from the second phosphate group of one strand forming a pentagonal arrangement with three water molecules, two of which form one side of a hexagon of second-shell waters. This is complemented by an equivalent pentagon from the other strand in the opposite direction, which forms the opposite side of this water hexagon. The minor groove is narrow at this point, with a width very close to that found in the crystal structures of oligonucleotides with narrow minor-groove A/T tracts (Fig. 3b). The arrangement of first-shell water molecules found here has some analogy with the spine of hydration found in these structures. Thus we see here that N2G and O2C of both terminal C:G base pair directly hydrogen bond to three water molecules, forming a short stretch of water spine. The symmetry of the solvent network is not exact; close inspection reveals several instances where hydrogen bonds on one side do not match those on the other.
Figure 3.
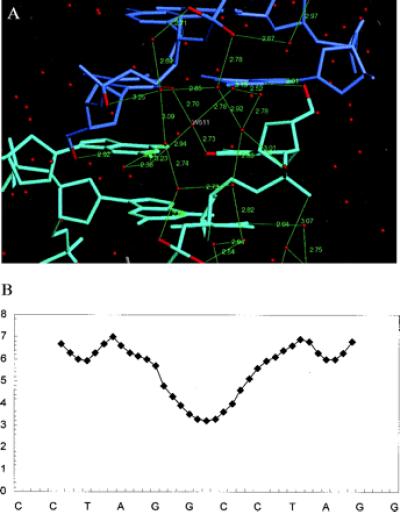
(a) The arrangement of DNA … water and water … water hydrogen bonds at the interface of the two hexamer duplexes, with hydrogen atoms shown in green. Note the water molecule W511 on the pseudo-2-fold axis. (b) Plot of minor-groove width (in angstroms), as ordinate, against the sequence as abscissa of the quasicontinuous stack of two hexanucleotide duplexes. The distances were calculated with the curves program (29).
The DNA Duplexes Have Local Distortions.
Both hexanucleotide duplexes in the asymmetric unit are overall in B-DNA conformations, although there are a number of features with considerable deviations from canonical B-DNA ideality. Thus the two TpA regions of the minor groove in the pseudo-dodecanucleotide have a wide (and deep) minor groove, with a maximum width of 6.8 Å (Fig. 3b), compared with 6.0 Å for canonical B-DNA. This exceptional width is required for minor-groove binding of the symmetry-equivalent TMPy molecule (Fig. 2 a and b). The groove progressively narrows toward the central quasicontinuous 5′-GG, 5′-CC segment of the pseudododecamer duplex, to a minimum width of 3.2 Å.
The A:T base pair have only modest propeller twist, of −10 to −13° (Table 2). The behavior of the two end 5′-terminal C:G base pair in each (approximately equivalent) hexamer duplex is highly unusual (Fig. 4). The terminal C:G of the dimer is propeller twisted by +28°, i.e., in the opposite direction to that observed in almost all other DNA structures. The adjacent C:G base pair is twisted by almost the same amount (−22°), but in the opposite conventional direction. These two 3′-terminal C:G base pair have high roll values, which are most clearly appreciated by considering each strand separately. The single-strand roll at the CpC step is very high, −34°, opening toward the major groove (Fig. 4), whereas that at the next step (CpT) is +19°, i.e., in the opposite direction. The opposite strand has a +20° roll at the terminal GpG step and one of 11° at the next GpA step. There are also appreciable perturbations in other base pair and base step morphological features, notably in buckle, slide, and opening, especially for those base pair close to the TMPy molecules (Table 2).
Table 2.
Base-pair and base-step morphological parameters, calculated with the curves program (28), for the two independent duplexes. These are listed as the pseudododecanucleotide duplex, and the two end base pairs are C1-G12 and G18-C19. Propeller twist, buckle, roll, and twist are in °, and slide is in angstroms.
| Pr twist | Buckle | ||
|---|---|---|---|
| Base pairs | |||
| Duplex 1 | |||
| C1-G12 | 29 | −12 | |
| C2-G11 | −22 | −11 | |
| T3-A10 | −10 | 5 | |
| A4-T9 | −13 | −1 | |
| G5-C8 | −17 | 19 | |
| G6-C7 | −28 | 3 | |
| Duplex 2 | |||
| C13-G24 | −28 | −5 | |
| C14-G23 | −12 | −18 | |
| T15-A22 | −13 | 2 | |
| A16-T21 | −11 | −3 | |
| G17-C20 | −22 | 11 | |
| G18-C19 | 24 | 15 | |
| Base steps | Roll | Twist | Slide |
| C1pC2 | −7 | 34 | −0.6 |
| C2pT3 | 16 | 28 | −0.1 |
| T3pA4 | −12 | 46 | 0.9 |
| A4pG5 | 15 | 24 | 0.5 |
| G5pG6 | −8 | 41 | 0.1 |
| G6pC13 | 2 | 46 | −1.9 |
| C13pC14 | −9 | 40 | 0.1 |
| C14pT15 | 16 | 24 | 0.5 |
| T15pA16 | −13 | 47 | 1 |
| A16pG17 | 16 | 27 | −0.1 |
| G17pG18 | −7 | 35 | −0.5 |
Figure 4.
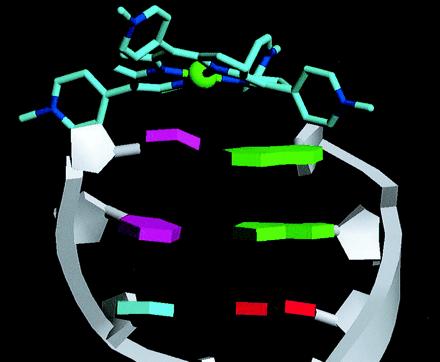
View of the end of one hexamer duplex with bases shown as idealized polygons and the end-capping Ni2+-TMPy ligand.
These marked distortions from normality are a consequence of the nonplanarity of the TMPy molecules, and indeed their nature and direction can be seen visually to complement that of the adjacent TMPy molecule (Fig. 4). Thus the concave buckle in the terminal C:G closely parallels the convex surface of the TMPy. The positions of the individual cytosine and guanine bases here are defined by their need to stack in a parallel manner with an individual pyrrole ring of the TMPy molecule. We conclude that the nonplanarity of the porphyrin is directly responsible for the distortions in base pair morphology.
The Porphyrin Geometry.
The highly nonplanar conformations for the two Ni2+-TMPy molecules (Figs. 2 a–c and 4) contrast remarkably with the planar conformations found for the native and Cu2+-TMPy structures (14, 15). Each pyrrole ring is closely planar, with a maximum deviation of 0.03 Å from the least-squares plane; however, each is twisted with respect to the next, with angles varying from 18 to 27°. The two pairs of pyrrole rings are twisted in the opposite direction, resulting in a saddle shape for the porphyrin. This type of deviation has been observed in the crystal structures of a number of substituted metalloporphyrins (23–26). The distortions are most likely to be a consequence of the short Ni-N(pyrrole) distances (1.91–1.95 Å), which result in shortening of the N … N distances involving opposite pyrroles, compared with those in metal-free TMPy. The larger Cu2+ ion [in its TMPy complex (15) with the hexanucleotide d(CGTACG)] has significantly longer Cu2+-N distances, of 2.03–2.11 Å, which enable the TMPy ligand here to assume a planar conformation. The nonplanarity of the Ni2+-TMPy molecules implies a diminution of the delocalization in the porphyrin π-bonding system and thus a decreased tendency for electron transfer within it. The four N-methyl-pyridyl substituents are located two in each groove, with the rings all oriented propeller like at angles of 55–65° to the porphyrin.
TMPy Groove Binding and Implications for DNA Strand Cleavage.
The TMPy molecule fits snugly into the A/T region of the minor groove, with the two inner N-methyl-pyridine groups oriented almost parallel to the groove floor (Fig. 2a). This orientation, which maximizes nonbonded contacts with the groove (Fig. 2 b and c), also explains in part the necessity for the groove to be wide at this point. It is notable that there are numerous close contacts between the TMPy molecule and the atoms forming the floor and walls of the groove surface (Fig. 2c), with 13 distances <3.5 Å. Some of these stabilizing contacts involve backbone atoms such as C4′, whereas others involve base substituents such as O2 of thymine. Several of the distances are short (≈3 Å) and imply the involvement of X … H-C hydrogen bonds.
The “carrier” duplex on which the groove-binding TMPy molecule is stacked (Fig. 2 a–c) appears to interact only weakly with the recipient groove. The recipient groove is slightly widened at this point as a result of the carrier duplex, even though there are very few close contacts between them, with the only intermolecular contact distances <3.5 Å between a pair of hydrogen bonds between the penultimate C:G base pair of the carrier and backbone oxygen atoms of the recipient duplex (Fig. 2c). The overall arrangement is undoubtedly held together by the strength of the porphyrin-groove contacts. We speculate that the absence of the carrier duplex could result in a slightly narrower groove at this point, although the widening is consistent with earlier reports of DNA distortion at the groove-binding site (2) and could be used to accommodate bulky axial substituents on a porphyrin molecule.
The center of the porphyrin, containing the nickel ion, is necessarily constrained to be close to the mouth of the groove. Significantly, the nickel ion is also close to one backbone and in particular to the C1′ and O3′ atoms of this backbone. This provides a plausible starting point for any subsequent photo-induced or oxygen-mediated strand cleavage (27). It is known that the latter occurs within the minor groove and involves oxidative or free-radical attack on deoxyribose sugars, especially at the C1′ position. We suggest that the observed proximity of the metal ion to them is a good model for the initial stage in this attack and explains why the sugars, rather than backbone phosphates, are cleaved.
The nonplanarity of the Ni2+-TMPy porphyrin suggests that it will not readily intercalate into duplex DNA without inducing significant structural perturbations, analogous to those produced in the immediate vicinity of the end-stacked porphyrin. The need for such distortions also suggests that the minor-groove interaction is the more energetically accessible binding mode for Ni2+-TMPy, by contrast with the ready intercalation of a rhodium–phenanthrene complex (28), even though it also requires the A/T minor groove to be widened. Similarly, the pseudointercalation of the Cu2+-TMPy ligand between CpG sites (15) requires this porphyrin to be planar.
Acknowledgments
This project was commenced while two of the authors (R.M. and M.S.) were working with S.N. at the Institute of Cancer Research. It was continued and brought to fruition in the laboratory of M.S. at King's College London. We are grateful to Professor David Wilson for providing samples of TMPy and for numerous discussions, to Drs. A. Thompson and G. Leonard for expert advice and assistance at the ESRF Beamline BM14, to Dr. Matthias Wilmann for his help at the Deutsches Elektronen Synchrotron, Hamburg Beamline X11, and to Mr. Tom Rutherford for excellent in-house x-ray technical assistance at King's College. The Cancer Research Campaign is thanked for support of studies on nucleic acid-ligand recognition (program grant SP1384 to S.N.), and we thank European Union Structural Biology Framework IV for a grant to M.S.
Abbreviations
- TMPy
tetra-(N-methylpyridyl) porphyrin
- Ni2+-TMPy
nickel2+-meso-tetra [4-N-methyl(pyridyl)] porphyrin
- MAD
multiwavelength anomalous diffraction
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160271897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160271897
References
- 1.Fiel R J. J Biomol Struct Dyn. 1989;6:1259–1274. doi: 10.1080/07391102.1989.10506549. [DOI] [PubMed] [Google Scholar]
- 2.Sehlstedt U, Kim S K, Carter P, Goodisman J, Vollano J F, Nordén B, Dabrowiak J C. Biochemistry. 1994;33:417–426. doi: 10.1021/bi00168a005. [DOI] [PubMed] [Google Scholar]
- 3.Dougherty T J. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villanueva A, Caggiari L, Jori G, Milanesi C. J Photochem Photobiol. 1994;B 23:49–56. doi: 10.1016/1011-1344(93)06982-9. [DOI] [PubMed] [Google Scholar]
- 5.Banville D L, Marzilli L G, Wilson W D. Biochem Biophys Res Commun. 1983;113:148–154. doi: 10.1016/0006-291x(83)90444-8. [DOI] [PubMed] [Google Scholar]
- 6.Bromley S D, Ward B W, Dabrowiak J C. Nucleic Acids Res. 1986;14:9133–9148. doi: 10.1093/nar/14.22.9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izbicka E, Wheelhouse R T, Raymond E, Davidson K K, Lawrence R A, Sun D, Windle B E, Hurley L H, Von Hoff D D. Cancer Res. 1999;59:639–644. [PubMed] [Google Scholar]
- 8.Wheelhouse R T, Sun D, Han H, Han F X, Hurley L H. J Am Chem Soc. 1998;120:3261–3262. [Google Scholar]
- 9.Han F X, Wheelhouse R T, Hurley L H. J Am Chem Soc. 1999;121:3561–3570. [Google Scholar]
- 10.Arthanari H, Basu S, Kawano T L, Bolton P H. Nucleic Acids Res. 1998;26:3724–3728. doi: 10.1093/nar/26.16.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ford K G, Fox K R, Neidle S, Waring M J. Nucleic Acids Res. 1987;15:2221–2234. doi: 10.1093/nar/15.5.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marzilli L G, Banville D L, Zon G, Wilson W D. J Am Chem Soc. 1986;108:4188–4192. [Google Scholar]
- 13.Guliaev A B, Leontis N B. Biochemistry. 1999;38:15425–15437. doi: 10.1021/bi9913808. [DOI] [PubMed] [Google Scholar]
- 14.Ford K G, Pearl L H, Neidle S. Nucleic Acids Res. 1987;15:6553–6562. doi: 10.1093/nar/15.16.6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipscomb L A, Zhou F X, Presnell S R, Woo R J, Peek M E, Plaskon R R, Williams L D. Biochemistry. 1996;35:2818–2823. doi: 10.1021/bi952443z. [DOI] [PubMed] [Google Scholar]
- 16.Ford K G, Neidle S. Bioorg Med Chem. 1995;3:671–677. doi: 10.1016/0968-0896(95)00052-i. [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 18.Bailey S. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 19.McRee D E. J Mol Graphics. 1992;10:44–47. [Google Scholar]
- 20.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:505–524. [PubMed] [Google Scholar]
- 21.Parkinson G, Vojtechovsky J, Clowney L, Brünger A T, Berman H M. Acta Crystallogr D. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 22.hyperchem, Ver. 5.01 (1997) (Hypercube, Gainesville, FL).
- 23.Medforth C J, Senge M O, Smith K M, Sparks L D, Shelnutt J A. J Am Chem Soc. 1992;114:9859–9869. [Google Scholar]
- 24.Barkigia K M, Renner M W, Furenlid L R, Medford C J, Smith K M, Fajer J. J Am Chem Soc. 1993;115:3627–3635. [Google Scholar]
- 25.Jentzen W, Simpson M C, Hobbs J D, Song X, Ema T, Nelson N Y, Medford C J, Smith K M, Veyrat M, Mazzanti M, et al. J Am Chem Soc. 1995;117:11085–11097. doi: 10.1021/ja00150a008. [DOI] [PubMed] [Google Scholar]
- 26.Medforth C J, Muzzi C M, Shea K M, Smith K M, Abraham R J, Jia S, Shelnutt J A. J Chem Soc Perkin Trans. 1997;2:833–837. [Google Scholar]
- 27.Mestre B, Jakobs A, Pratviel G, Meunier B. Biochemistry. 1996;35:9140–9149. doi: 10.1021/bi9530402. [DOI] [PubMed] [Google Scholar]
- 28.Kielkopf C L, Erkkila K E, Hudson B P, Barton J K, Rees D C. Nat Struct Biol. 2000;7:117–121. doi: 10.1038/72385. [DOI] [PubMed] [Google Scholar]
- 29.Lavery R, Sklenar H. J Biomol Struct Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]