

Abstract
A major hallmark of apoptosis is normotonic shrinkage of cells. Here, we studied the relation between apoptotic cell shrinkage and apoptotic cell death. Induction of the apoptotic volume decrease (AVD) under normotonic conditions was found to be coupled to facilitation of the regulatory volume decrease (RVD), which is known to be attained by parallel operation of Cl− and K+ channels, under hypotonic conditions. Both the AVD induction and the RVD facilitation were found to precede cytochrome c release, caspase-3 activation, DNA laddering, and ultrastructural alterations in three cell types after apoptotic insults with two distinct apoptosis inducers. Also, the AVD was not prevented by a broad-spectrum caspase inhibitor. When the AVD induction and the RVD facilitation were prevented by blocking volume-regulatory Cl− or K+ channels, these cells did not show succeeding apoptotic biochemical and morphological events and were rescued from death. Thus, it is concluded that the AVD, which is caused by disordered cell volume regulation, is an early prerequisite to apoptotic events leading to cell death.
Apoptosis, which is a morphologically and biochemically defined form of cell death, occurs in response to a variety of stimuli under physiological and pathological circumstances. A major hallmark of apoptosis is normotonic shrinkage of cells (1). The apoptotic volume decrease (AVD), which starts before cell fragmentation, is coupled to K+ release from the cells (2–4), presumably via K+ channels (5–7). To drive the net efflux of water, which leads to cell shrinkage, release of anions should take place in parallel with K+ release because of the restraint of electroneutrality. Stimulation of CD95 (Fas) receptor has actually been shown to induce activation of outwardly rectifying Cl− channels (8), which were also activated by osmotic swelling (9), in lymphoid Jurkat cells. Thus, there exists a possibility that volume regulation mechanisms (10–12), including volume-regulatory Cl− and K+ channels, are disordered, thereby inducing the AVD during the apoptotic process. Because Bortner and Cidlowski (13) previously reported that persistent physical shrinkage induced by hyperosmotic stress leads to apoptosis in lymphoid cells, it is also possible that the AVD is a prerequisite to apoptotic cell death. These possibilities were here examined in human epithelial HeLa, human lymphoid U937, and mouse neuroblastoma × rat glioma hybrid NG108-15 and rat pheochromocytoma PC12 cells by applying a mitochondrial apoptotic inducer, staurosporine (STS), or a receptor-mediated apoptotic inducer, tumor necrosis factor α plus cycloheximide (TNF/CHX).
Materials and Methods
Cell Culture and Apoptosis Induction.
HeLa and U937 cells were cultured in MEM and RPMI 1640 media, respectively, supplemented with 10% FBS. PC12 and NG108-15 cells were cultured in DMEM plus 10% FBS and provided for experiments without inducing neuronal differentiation.
To induce apoptosis, these cells in the log-growing phase were treated with STS (1, 4, 4, and 8 μM for U937, HeLa, NG108-15, and PC12 cells, respectively), as previously described (14), or TNFα (Endogen, Woburn, MA; 2, 10, and 10 ng ml−1 for U937, PC12, and NG108-15 cells, respectively) plus CHX (0.1, 1, and 1 μg ml−1 for U937, PC12, and NG108-15 cells, respectively), as previously reported (15).
Cell Volume Measurement.
Cell volume was measured by an electronic sizing technique with a Coulter-type cell size analyzer (CDA-500; Sysmex, Kobe, Japan), as previously described (16). The mean volume of the population was calculated by a computer from the cell volume distribution based on those of latex beads with known volume. Isotonic or hypotonic solution consisted of (mM) 95 NaCl, 4.5 KCl, 1 MgCl2, 1 CaCl2, 110, or 0 mannitol, and 5 Hepes/NaOH (pH 7.3; 310 or 200 mosmol kg⋅H2O−1).
Cytochrome c Release Assay.
Loss of cytochrome c from mitochondria was observed by immunostaining under a confocal, laser scanning fluorescence microscope (Bio-Rad MRC-1024), as previously reported (17), using an anti-cytochrome c monoclonal antibody (6H2.B4; PharMingen). Propidium iodide was used as a counterstain. Cytochrome c release to cytosol was monitored by Western blot analysis using an anti-cytochrome c monoclonal antibody (7H8.2C12; PharMingen) for cytosolic fractions, as previously described (18).
Caspase-3 Activity Measurements.
Caspase-3 activity was measured by using a fluorometric assay (19). To exclude an involvement of other related proteases, the difference between fluorescence in the absence and presence of the specific inhibitor of caspase-3 was observed. The fluorogenic substrate, which was labeled with the fluorochrome 7-amino 4-methyl coumarin (AMC), for caspase-3 (Ac-DEVD-AMC) and the tetrapeptide inhibitor of caspase-3 (Ac-DEVD-CHO) were provided in the CaspASE Assay System (Promega).
DNA Fragmentation Assay.
Internucleosomal DNA fragmentation was detected by DNA ladder, as previously described (20). Briefly, cells were digested in lysis buffer (10 mM EDTA/0.5% Na-N-lauroylsarcosinate/500 μg ml−1 RnASE/50 mM Tris⋅HCl, pH 7.8) at 37°C for 1 h and treated with 500 μg ml−1 proteinase K at 37°C for 1 h. The chromosomal DNA was analyzed by agarose gel electrophoresis (2%), followed by staining with ethidium bromide.
Transmission Electron Microscopy.
Electron microscopical observations were made by a JEM 100CX (Tokyo, Japan). Cell cultures were prefixed by modified Karnovsky fixative (21) using 0.1 M Na-phosphate buffer without adding CaCl2. After postosmification with 1% OsO4 in water, cells were dehydrated in a graded ethanol series, and embedded in Epon.
Cell Viability Assay.
Viability of cells cultured in the 96-well culture plates was assessed by mitochondrial dehydrogenase activity using the colorimetric MTT assay, based on the fact that viable cells (but not dead cells) can reduce 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (22), using the Cell Counting Kit (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions. Cell viability was also assessed by trypan blue exclusion after a 5-min incubation with 0.4% trypan blue.
Results
AVD Induction and Its Sensitivity to Channel Blockers.
Treatment for 2 h with the bacterial alkaloid STS resulted in significant reduction of mean cell volume in epithelial HeLa and lymphoid U937 (Fig. 1A) as well as in neuronal NG108-15 and PC12 cells (data not shown; n = 20). Application (for 2 h) of TNF/CHX also induced shrinkage in NG108-15 and U937 cells (Fig. 1A) as well as in HeLa and PC12 cells (data not shown; n = 20). Within 3 h after apoptogenic stimulation, cell volume distribution exhibited no additional population with smaller cell size because of apoptotic body formation, and electron microscopic studies indicated that no cell fragmentation started (data not shown).
Figure 1.
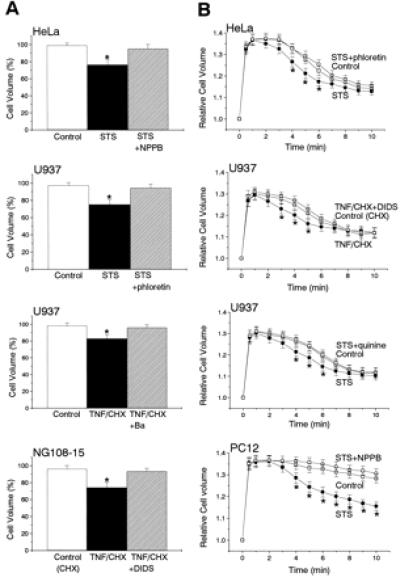
Induction of AVD (A) and facilitation of RVD (B) by the 2-h treatment with STS or TNF/CHX, and their prevention by simultaneous treatment with a channel blocker, 0.5 mM NPPB, 30 μM phloretin, 0.5 mM DIDS, 5 mM Ba2+, or 0.5 mM quinine in U937, HeLa, NG108-15, and PC12 cells. Cell volume measurements were performed in the absence of any drugs after washing out apoptotic inducers and channel blockers, and the data were normalized by those measured immediately before the 2-h treatment (A) or a hypotonic challenge (B). Each data represents the mean ± SEM (vertical bar) of 16 observations. *, P < 0.05 vs. corresponding control.
This early-phase cell shrinkage associated with apoptosis, termed AVD, was completely inhibited by a Cl− channel blocker, 5-nitro-2-(3-phenylpropylamino)-benzoate (NPPB) or 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) (0.5 mM; Fig. 1A). Other Cl− channel blockers, 4-acetamido-4′-isothiocyanostilbene (SITS), niflumic acid, and glibenclamide, were also effective (0.5 mM; data not shown; n = 10 each). Because these chemicals are commonly known to block volume-sensitive Cl− channels (12), which are activated by cell swelling in a large variety of cell types, we tested another drug, phloretin, which has been recently shown to block, at low concentrations (below 100 μM), volume-sensitive Cl− channels but not cAMP- or Ca2+-activated Cl− channels in epithelial cells (23). Phloretin (30 μM) could also prevent the AVD induced by STS (Fig. 1A) and by TNF/CHX in U937 cells as well as by either apoptotic inducer in three other cell types (data not shown; n = 10). A known blocker of volume-regulatory K+ channels (10), Ba2+ (5 mM; Fig. 1A) or quinine (0.5 mM; data not shown; n = 10), also abolished the AVD in U937 cells (Fig. 1A) and three other cell types treated with STS or TNF/CHX (data not shown; n = 10 each).
Regulatory Volume Decrease (RVD) Facilitation and Its Block by Pretreatment with Channel Blockers.
HeLa and U937 cells could respond with osmotic swelling to a hypotonic challenge (65% osmolality) and thereafter exhibited a RVD (Fig. 1B, □), irrespective of treatment with CHX. The RVD is known to be attained by activation of both K+ and Cl− conductances in most mammalian cell types (10, 11). Actually, the RVD was abolished in these cells when a blocker of volume-sensitive Cl− channels (0.5 mM DIDS, 0.5 mM NPPB, or 30 μM phloretin) or K+ channels (5 mM Ba2+ or 0.5 mM quinine) was added during cell volume measurements (data not shown; n = 16 each).
HeLa and U937 cells undergoing apoptosis (2 h after induction) also responded with swelling and RVD to hypotonic stress, but the RVD time course in these apoptotic cells was found to be hastened (Fig. 1B, ●), compared with that in control cells (□). Control PC12 cells showed little RVD within 10 min (Fig. 1B Bottom, □). Two hours after induction of apoptosis, however, PC12 cells exhibited distinct RVD (●). Similar observations were also obtained in NG108-15 cells (data not shown; n = 16). Such facilitation of the RVD response was never observed in these cells when phloretin, DIDS, or NPPB had been simultaneously applied during application of an apoptotic inducer and then removed immediately before cell volume measurements (Fig. 1B, ○). Similar effects were also observed by pretreatment with another volume-sensitive Cl− channel blocker, SITS or glibenclamide (0.5 mM; data not shown; n = 10 each) and with a K+ channel blocker, quinine (0.5 mM; Fig. 1B) or Ba2+ (5 mM; data not shown; n = 10). These results indicate that stimulation with an apoptotic inducer may somehow, even under normotonic conditions, lead to activation of volume-regulatory Cl− and K+ channels that can usually partake in the RVD process under hypotonic conditions.
Cytochrome c Release and Its Block by Channel Blockers.
Both immunocytochemistry and Western blot demonstrated that either 4- to 8-h application of STS or TNF/CHX induces release of cytochrome c from mitochondria in HeLa (Fig. 2A) and to cytosol in HeLa, U937, and PC12 cells (Fig. 2B). Apoptotic cytochrome c release was prevented by a Cl− channel blocker, DIDS, NPPB, or phloretin. There may exist a possibility that Cl− channel blockers inhibited mitochondrial VDAC/porin anion channels, which are likely to mediate cytochrome c release (24, 25). However, a K+ channel blocker, quinine (Fig. 2B) or Ba2+ (5 mM; data not shown; n = 3), was also effective to suppress STS-induced cytochrome c release.
Figure 2.
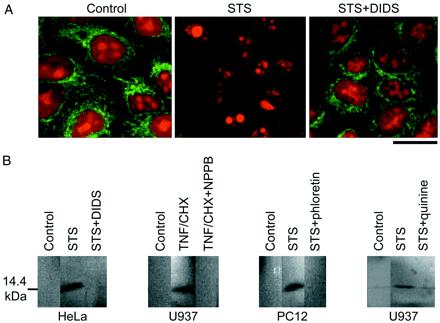
Release of cytochrome c induced by treatment with an apoptogenic inducer, and its prevention by simultaneous treatment with a Cl− or K+ channel blocker. (A) Cytochrome c release from mitochondria monitored by immunocytochemistry in HeLa cells 5.5 h after treatment with STS in the absence or presence of 0.5 mM DIDS. A green fluorescence: mitochondrial cytochrome c revealed by FITC-conjugated secondary antibody. A red fluorescence: counter staining with propidium iodide. Data represent triplicate experiments. (Scale: 20 μm). (B) Cytochrome c release to cytosol was monitored by Western blot in U937, HeLa, and PC12 cells 8 h after treatment with STS or TNF/CHX in the absence or presence of 0.5 mM DIDS, 0.5 mM NPPB, 30 μM phloretin, or 0.5 mM quinine. Data represent triplicate experiments.
Caspase Activation, DNA Laddering, and Their Prevention by Channel Blockers.
Activation of caspase-3 and laddering of DNA were evoked by 4- to 8-h application of STS or TNF/CHX in all of the cell types used, as shown in Fig. 3 A and B, respectively. These two biochemical signs indicative of apoptosis, however, were abolished by simultaneous application of DIDS, NPPB, or phloretin (Fig. 3), or of SITS (data not shown; n = 4) with either apoptotic inducer, STS or TNF/CHX. A K+ channel blocker, Ba2+ (Fig. 3) or quinine (0.5 mM; data not shown; n = 4), also prevented STS- or TNF/CHX-induced activation of caspase-3 and laddering of DNA in U937 cells (Fig. 3) as well as other cell types (data not shown; n = 4 each).
Figure 3.
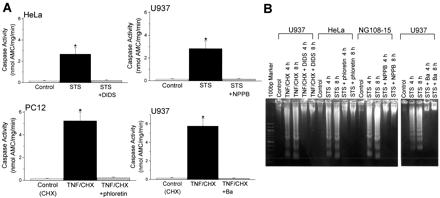
Caspase-3 activation (A) and DNA laddering (B) induced by the 4-h or 8-h treatment with STS or TNF/CHX, and their prevention by simultaneous treatment with 0.5 mM DIDS, 0.5 mM NPPB, 30 μM phloretin, or 5 mM Ba2+ in U937, HeLa, PC12, and NG108-15 cells. In A, each column represents the mean ± SEM (vertical bar) of six observations. Data in B are representative of triplicate experiments. *, P < 0.05 vs. control.
Apoptotic Morphology and Its Prevention by Channel Blockers.
As demonstrated in Fig. 4 A and B, treatment of HeLa, U937, and PC12 cells with STS or TNF/CHX induced ultrastructural alterations characteristic of apoptosis (1), such as chromatin condensation at the periphery of the nucleus, leaky nuclear envelopes, and intracellular vacuolation within 4 h, as gauged by transmission electron microscopy. These morphological signs of apoptosis were largely abolished by simultaneous treatment with a Cl− channel blocker, DIDS, NPPB, or phloretin (Fig. 4C). Also, a K+ channel blocker (5 mM Ba2+ or 0.5 mM quinine) was found to be effective in blocking the morphological changes induced by TNF/CHX in PC12 cells (data not shown).
Figure 4.
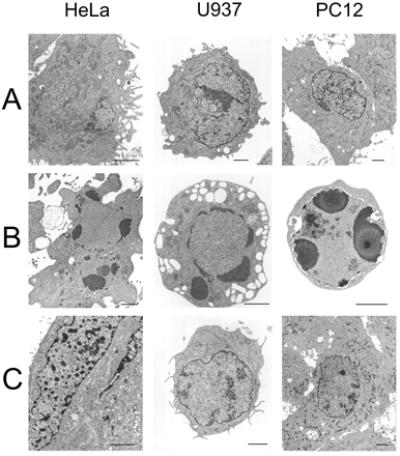
Changes in morphological features under thin section electron microscopy 4 h after treatment with STS or TNF/CHX, and their prevention by simultaneous treatment with 0.5 mM NPPB, 0.5 mM DIDS, and 30 μM phloretin in HeLa (Left), U937 (Center), and PC12 (Right) cells, respectively. (Scale: 2 μm.) (A) Control HeLa, U937, and PC12 cells. (B) HeLa and U937 cells 4 h after treatment with STS, and PC12 cells 4 h after treatment with TNF/CHX. (C) These cells 4 h after treatment with STS (Left and Center) or TNF/CHX (Right) simultaneously with 0.5 mM NPPB (Left), 0.5 mM DIDS (Center), or 30 μM phloretin (Right).
Apoptotic Cell Death and Its Prevention by Channel Blockers.
Treatment for 8 h with STS resulted in marked reduction of cell survival assessed by the MTT assay in HeLa, U937, PC12 (Fig. 5), and NG108-15 cells (data not shown; n = 20). Application of TNF/CHX (for 8 h) also induced cell death in U937 (Fig. 5), HeLa, and PC12 cells (data not shown; n = 20 each). Simultaneous application of a known Cl− channel blocker, NPPB or DIDS, was found to prevent cell death in HeLa and PC12 cells (Fig. 5) as well as in U937 and NG108-15 cells (data not shown; n = 20 each). Other Cl− channel blockers, SITS, niflumic acid, and glibenclamide, were also effective (0.5 mM; data not shown; n = 10 each). Phloretin could also prevent TNF/CHX-induced death of U937 (Fig. 5) as well as STS-induced death of HeLa, U937, and NG108-15 cells (data not shown; n = 20 each) at 30 μM. Apoptotic cell death was blocked by a K+ channel blocker, Ba2+ (5 mM), in STS-treated U937 cells (Fig. 5) as well as in TNF/CHX-treated U937 and STS-treated HeLa cells (data not shown; n = 20 each). Another K+ channel blocker, quinine (0.5 mM), also prevented STS-induced cell death in NG108-15 cells (data not shown; n = 20). Essentially the same results were obtained when cell survival was assessed by trypan blue exclusion in U937 cells (see Fig. 6C) as well as in HeLa, PC12, and NG108-15 cells (data not shown; n = 12 each). In contrast, neither anthracene-9-carboxylate (1 mM), which is a known blocker of cAMP-activated (CFTR) Cl− channels, nor furosemide (0.5 mM), which blocks not only Na+-K+-2Cl− or Na+-Cl− symporters but also epithelial Ca2+-activated Cl− channels (26), prevented STS-induced death of HeLa and U937 cells (data not shown; n = 10 each for MTT assay and n = 4 each for trypan blue assay).
Figure 5.
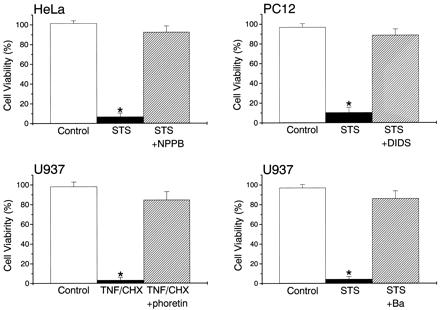
Cell death induced by an 8-h treatment with STS or TNF/CHX, and its rescue by simultaneous treatment with 0.5 mM NPPB, 0.5 mM DIDS, 30 μM phloretin, or 5 mM Ba2+ in HeLa, U937, and PC12 cells. Cell viability was assessed by the MTT assay, and the data were normalized by those measured immediately before the 8-h treatment. Each column represents the mean ± SEM (vertical bar) of 20 observations. Control data for STS experiments were obtained after an 8-h incubation without adding any drugs, and those for TNF/CHX experiments were after 8-h treatment with CHX alone. *, P < 0.05 vs. control.
Figure 6.
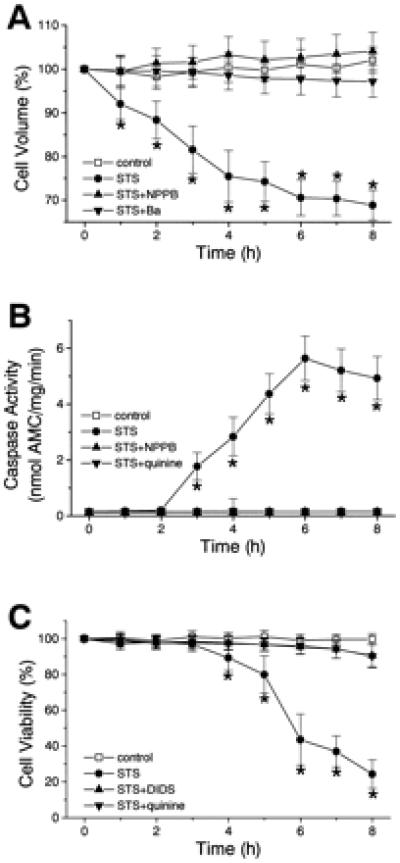
The time courses of changes in the mean cell volume (A), caspase-3 activity (B), and cell viability (C) in control (□) and STS-induced apoptotic cells (filled symbols) in the absence (●) or presence of a Cl− channel blocker (▴) or of a K+ channel blocker (▾). After a 4-h treatment with STS, bimodal cell volume distribution was observed presumably because of formation of apoptotic body. Cell viability was assessed by trypan blue exclusion. *, P < 0.05 vs. control.
AVD Induction Before Known Biochemical Apoptotic Events.
The time courses of AVD, biochemical apoptotic events, and cell death induced by STS were compared in U937 cells. As shown in Fig. 6 (●), cell shrinkage started as early as 1 h after application of STS (A), whereas caspase-3 activation (B) and cell death (C) were not observed within 2 h and 3 h after STS treatment, respectively. Neither cytochrome c release nor DNA laddering was observed within 2 h after stimulation with STS in all cell types tested (data not shown; n =4 each). Even when apoptotic cell death, caspase-3 activation, and DNA laddering were prevented by a broad-spectrum caspase inhibitor, zVAD-fmk (50 μM), the AVD was normally observed in STS-treated U937 cells. The mean U937 cell volume decreased to 90.2 ± 4.1% and 88.3 ± 3.1% (n = 4) 2 h after application of STS with and without zVAD-fmk, respectively. This is in good agreement with previous observations in Jurkat cells treated with A23187 or thapsigargin (27). Similar results were also observed with another caspase inhibitor, zD-dcb (0.1 mM; data not shown; n =6).
When stimulated with TNF/CHX, cell shrinkage was found to start within 30 min, whereas cytochrome c release, caspase-3 activation, and DNA laddering were never observed within 30 min in U937 cells (data not shown; n = 4–12). Taken together, it appears that the AVD is an event upstream to cytochrome c release, caspase activation, DNA laddering, and cell death.
Discussion
All of the apoptotic events examined in the present study were found to be prevented by NPPB, DIDS, SITS, niflumic acid, glibencamide, and phloretin, which are known as blockers of volume-regulatory Cl− channels (12, 23). Similarly, previous studies showed that, at a much higher concentration (2 mM), DIDS exhibited an inhibitory effect on an STS-induced DNA fragmentation monitored by terminal dUTP nick-end labeling in rat cerebellar granule neurons (28), and also that a low concentration of glibenclamide (0.1 mM), DIDS (0.1 mM), IAA (0.1 mM), or DPC (1 mM), although only in part, reduced DNA fragmentation and annexin V binding in Fas antigen-treated Jurkat lymphoid cells (8). These drugs are known to be effective blockers of volume-sensitive Cl− channels (12). In contrast, blockers of cAMP-activated (CFTR) Cl− channels and that of epithelial Ca2+-activated Cl− channels were found to be ineffective. These results suggest that volume-regulatory Cl− channels are involved in the AVD. In fact, the cells exhibiting the AVD responded to a hypotonic challenge with a faster RVD, which is known to involve volume-regulatory Cl− channels. In addition, Szabo et al. (8) have recently shown that stimulation of Fas receptors induced activation of outwardly rectifying Cl− channels in lymphoid Jurkat cells. Furthermore, our preliminary study using a Cl−-sensitive fluorescent dye (MQAE) showed that a significant decrease in the intracellular Cl− concentration was observed within 1–2 h after treatment with STS in HeLa cells, and the apoptotic Cl− egress was abolished by NPPB (29). Taken together, it is concluded that apoptotic cell death can be rescued by preventing the early-phase apoptotic cell shrinkage (AVD) by blocking the volume-regulatory Cl− channel in lymphoid, epithelial, and neuronal cells.
Ba2+ and quinine, which are known blockers of volume-regulatory K+ channels (10), also prevented all of the apoptotic events examined in the present study. This is in good agreement with previous observations that another K+ channel blocker, TEA or 4-aminopyridine, blocked apoptotic cell death in other cell types (5–7). Thus, it is possible that some K+ channel is activated in apoptotic cells. In fact, up-regulation of voltage-dependent K+ channels was observed in neuronal and myelobastic cells undergoing apoptosis (5–7). Thus, it appears that the K+ channel activity is implicated in the AVD in concert with the Cl− channel activity.
Induction of AVD began within 0.5–2 h after apoptogenic stimulation and thus preceded cytochrome c release, caspase-3 activation, and DNA laddering. A broad-spectrum inhibitor of caspase, zVAD-fmk or zD-dcb, failed to prevent the AVD induction. Thus, it appears that the AVD is an upstream event of these biochemical apoptotic events. In fact, prevention of AVD by blockers of Cl− or K+ channels abolished these biochemical apoptotic events and ultrastructural alterations characteristic of apoptosis. Although its mechanism remains to be elucidated, it is noted that physical shrinkage itself was reported to induce apoptosis in lymphoid cells (13) and activation of mitogen-activated protein kinases (30), which are possibly involved in the apoptosis signaling (31–33). Also, a decrease in the intracellular K+ concentration, which is coincident with apoptotic cell shrinkage (25), was shown to be necessary for apoptotic cell death (34).
In summary, the present study indicated that the AVD is an early prerequisite to apoptotic cell death. It was also demonstrated that normotonic induction of AVD is coupled to the RVD facilitation under hypotonic conditions presumably because of excessive activation of volume-regulatory Cl− and/or K+ channels. Thus, the disordered cell volume regulation may provide a new target for cytoprotective drugs against apoptotic cell death associated with pathological insults.
Acknowledgments
We thank A. F. James for reading the manuscript, M. Ohara, A. Hattori, K. Shigemoto, and S. Tanaka for technical assistance, and K. Dezaki, Y. Ando-Akatsuka, S. Morishima, and R. Z. Sabirov for helpful discussions.
Abbreviations
- AVD
apoptotic volume decrease
- RVD
regulatory volume decrease
- STS
staurosporine
- TNF
tumor necrosis factor α
- CHX
cycloheximide
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NPPB
5-nitro-2-(3-phenylpropylamino)-benzoate
- DIDS
4,4'-diisothiocyanostilbene-2,2'-disulfonic acid
- SITS
4-acetamido-4'-isothiocyanostilbene
Footnotes
See commentary on page 9360.
References
- 1.Wyllie A H, Kerr J F R, Currie A R. Int Rev Cytol. 1980;68:251–307. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 2.Barbiero G, Duranti F, Bonelli G, Amenta J S, Baccino F M. Exp Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- 3.Benson R S P, Heer S, Dive C, Watson A J M. Am J Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- 4.Bortner C D, Hughes F M, Jr, Cidlowski J A. J Biol Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 5.Yu S P, Yeh C-H, Sensi S L, Gwag B J, Canzoniero L M T, Farhangrazi Z S, Ying H S, Tian M, Dugan L L, Choi D W. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Xu D, Dai W, Lu L. J Biol Chem. 1999;274:3678–3685. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- 7.Colom L V, Diaz M E, Beers D R, Neely A, Xie W-J, Appel S H. J Neurochem. 1998;70:1925–1934. doi: 10.1046/j.1471-4159.1998.70051925.x. [DOI] [PubMed] [Google Scholar]
- 8.Szabo I, Lepple-Wienhues A, Kaba K N, Zoratti M, Gulbins E, Lang F. Proc Natl Acad Sci USA. 1998;95:6169–6174. doi: 10.1073/pnas.95.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lepple-Wienhues A, Szabo I, Laun T, Kaba K N, Gulbins E, Lang F. J Cell Biol. 1998;141:281–286. doi: 10.1083/jcb.141.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okada Y, Hazama A. News Physiol Sci. 1989;4:238–242. [Google Scholar]
- 11.Hoffmann E K, Simonsen L O. Physiol Rev. 1989;69:315–382. doi: 10.1152/physrev.1989.69.2.315. [DOI] [PubMed] [Google Scholar]
- 12.Okada Y. Am J Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- 13.Bortner C D, Cidlowski J A. Am J Physiol. 1996;271:C950–C961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- 14.Ishizaki Y, Voyvodic J T, Burne J F, Raff M C. J Cell Biol. 1993;121:899–908. doi: 10.1083/jcb.121.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White E, Sabbatini P, Debbas M, Wold W S, Kusher D I, Gooding L R. Mol Cell Biol. 1992;12:2570–2580. doi: 10.1128/mcb.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazama A, Okada Y. J Physiol. 1988;402:687–702. doi: 10.1113/jphysiol.1988.sp017229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshmukh M, Johnson E M., Jr Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Kim C N, Yang J, Jemmerson R, Wang X. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 19.Thornberry N A. Methods Enzymol. 1994;244:615–631. doi: 10.1016/0076-6879(94)44045-x. [DOI] [PubMed] [Google Scholar]
- 20.Shiokawa D, Ohyama H, Yamada T, Takahashi K, Tanuma S. Eur J Biochem. 1994;226:23–30. doi: 10.1111/j.1432-1033.1994.tb20022.x. [DOI] [PubMed] [Google Scholar]
- 21.Karnovsky M J. J Cell Biol. 1965;27:137A. [Google Scholar]
- 22.Mossman T. J Immunol Methods. 1983;65:55–63. [Google Scholar]
- 23.Fan H T, Kida H, Morishima S, Oiki S, Okada Y. Jpn J Physiol. 1999;49,Suppl.:S109. [Google Scholar]
- 24.Green D R, Reed J C. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 25.Tsujimoto Y, Shimizu S. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- 26.Evans M G, Marty A, Tan Y P, Trautmann A. Pflügers Arch. 1986;406:65–68. doi: 10.1007/BF00582955. [DOI] [PubMed] [Google Scholar]
- 27.Bortner C D, Cidlowski J A. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- 28.Himi T, Ishizaki Y, Murota S. Annu Meet Soc Neurosci. 1995;21:812. [Google Scholar]
- 29.Dezaki, K., Maeno, E. & Okada, Y. (2000) Jpn. J. Physiol.50, Suppl., in press. [DOI] [PubMed]
- 30.Roger F, Martin P-Y, Rousselot M, Favre H, Feraille E. J Biol Chem. 1999;274:34103–34110. doi: 10.1074/jbc.274.48.34103. [DOI] [PubMed] [Google Scholar]
- 31.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matusmoto K, Miyazono K, Gotoh Y. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 32.Ashkenazi A, Dixit V M. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 33.Gotoh Y, Cooper J A. J Biol Chem. 1998;273:17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- 34.Hughes F M, Jr, Bortner C D, Purdy G D, Cidlowski J A. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]