

Abstract
c-Myc plays a vital role in cell-cycle progression. Deregulated expression of c-Myc can overcome cell-cycle arrest in order to promote cellular proliferation. Transforming growth factor β (TGFβ) treatment of immortalized human keratinocyte cells inhibits cell-cycle progression and is characterized by down-regulation of c-Myc followed by up-regulation of p21CIP1. A direct role of c-Myc in this pathway was demonstrated by the observation that ectopic expression of c-Myc overcame the cell-cycle block induced by TGFβ treatment. The induction of p21CIP1 transcription by TGFβ was blocked in human keratinocyte cells stably expressing c-Myc. Furthermore, overexpression of c-Myc in NIH 3T3 cells repressed the basal levels of p21CIP1 mRNA. Repression of p21CIP1 transcription by c-Myc occurred at the promoter level in a region near the start site of transcriptional initiation and was independent of histone deacetylase activity. These data suggest that the down-regulation of c-Myc after TGFβ signaling is important for subsequent regulation of p21CIP1 and cell-cycle inhibition. Thus, repression of the cell-cycle inhibitory gene p21CIP1 plays a role in c-Myc-dependent cell-cycle progression.
The c-myc protooncogene plays a critical role in cellular proliferation. Its deregulated expression leads to the development of cancer and homozygous deletion of c-myc in Rat1 fibroblasts leads to significant lengthening of the cell cycle (1). It has been demonstrated that ectopic expression of c-Myc promotes cell-cycle progression and shortens G1 phase in cycling cells (2, 3). The mechanism(s) by which c-Myc regulates the cell cycle is not fully understood, although c-Myc has been found to transactivate and transrepress several cell-cycle-regulatory genes (4–10).
Transforming growth factor β (TGFβ) treatment leads to G1 cell-cycle arrest of several cell types, including epithelial, endothelial, and hematopoietic cells. Loss of TGFβ responsiveness occurs in several types of cancer. This loss of responsiveness is typically the result of loss of TGFβ receptor expression or defects in downstream signaling events controlled by TGFβ (11–15). Inhibition of the cell cycle by TGFβ is thought to be mediated in part by down-regulation of proliferative proteins, such as c-myc, coupled with up-regulation of cell-cycle-inhibitory proteins, such as p15INK4b, p21CIP1, or p27KIP. Regulation of these genes by TGFβ signaling occurs at transcriptional and posttranslational levels (13, 16).
Increased expression of p21CIP1 is associated with cell-cycle inhibition, differentiation, and cellular senescence (17–19). The p21CIP1 protein is a member of a group of cell-cycle-inhibitory proteins including p27KIP1 and p57KIP2. These proteins function by associating with cyclin/cdk complexes to inhibit their activity. p21CIP1 transcription is regulated by several factors, including p53, C/EBP α and β, and E1A (20–23). Interestingly, p21CIP1 transcription is stimulated by cycloheximide treatment (24), implying transcription may be repressed by a protein factor with a short turnover rate.
It has been shown previously that ectopic expression of c-Myc will inhibit the action of TGFβ on the cell cycle in BALB/MK murine keratinocytes, M1 murine myeloid leukemia cells, and CCL64 mink-lung epithelial cells (25–27). We demonstrate here that ectopic expression of c-Myc also inhibited TGFβ-mediated repression of cell-cycle progression in human immortalized keratinocytes (HaCaT). This inhibition of TGFβ activity by c-Myc resulted in a repression of induction of p21CIP1 transcription. Furthermore, overexpression of c-Myc in NIH 3T3 murine fibroblasts led to repression of basal levels of p21CIP1 transcripts. We demonstrate that c-Myc repressed transcription of p21CIP1 at the promoter level, independently of histone deacetylase activity. The region of the p21CIP1 promoter repressed by c-Myc does not overlap with the TGFβ-responsive element or other previously identified regulatory regions but is in close proximity to the site of transcriptional initiation.
Materials and Methods
Antibodies and Reagents.
Generation and purification of anti-Mycfl and anti-av-Myc 12C antibodies have been described previously (28, 29). Recombinant human TGFβ1 was purchased from R & D Systems. Puromycin and hygromycin B were purchased from Calbiochem. Trichostatin A (TSA) was purchased from Sigma.
Plasmids.
A vector encoding the murine ecotropic virus receptor (hygro muEcoR) was obtained from Scott Lowe (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). Construction of CMV-Myc2 and pBABE-Myc2 has been previously described (30, 31). The murine c-Myc cDNA used in these constructs has a mutation at the CUG upstream initiation site to prevent synthesis of the c-Myc-1 protein (30). In addition, the murine penultimate carboxyl-terminal glycine was mutated to arginine so that the avian-specific antiserum (anti-av-myc 12C) can selectively immunoprecipitate the exogenous murine protein (31). A reporter vector containing a 2.3-kb fragment of the p21CIP1 promoter was obtained from Rebecca Chinery (Mayo Clinic) and has been described previously (22, 32). The p21CIP1 promoter deletion luciferase vectors p21P Sma, p21P SmaΔ1, and p21P SmaΔ2 were obtained from Xiao-Fan Wang (Duke University, Durham, NC) and have been described previously (33). The gadd45 reporter vector was obtained from Linda Penn (University of Toronto) and has been described previously (34). The human matrilysin promoter reporter vector was obtained from Howard Crawford (Vanderbilt University, Nashville, TN) and was made by subcloning a 2.3-kb Mfe I fragment of the human matrilysin promoter (35) into the EcoRI site of pGL2-Basic (Promega).
Transient Transfections.
For transient transfection assays, NIH 3T3 cells were transfected by using LipofectAmine (GIBCO/BRL) in serum-free media for 6 hr, then media were replaced with DMEM with 10% CS. Cells were harvested 48 hr after transfection. Relative levels of p21CIP1 mRNA were normalized to cyclophilin levels, and levels of p21CIP1 in vector-only-transfected cells were set to a value of one for comparison.
Generation of Stable Cell Lines.
HaCaT cells were maintained in DMEM with 10% FBS. To facilitate the infection of these human cells with an ecotropic murine retrovirus, HaCaTs were first stably infected with hygro muEcoR. Cells were selected with hygromycin at 500 μg/ml for approximately 7 days. Cells were then infected with viral supernatants containing pBABE empty vector or pBABE-Myc2 collected from ψ2 packaging cell lines. Cells were selected with puromycin at 1 μg/ml for approximately 3 days, and clonal cell lines were isolated.
For stable luciferase reporter cell lines, transfections were performed as described above. NIH 3T3 cells were cotransfected with full-length p21CIP1 promoter luciferase construct and empty pBABE-hygro. Cells were selected for 2 days with 500 μg/ml hygromycin B, then several clones were isolated. Individual clones were analyzed for basal luciferase activity, and four independent clones were subsequently infected with either empty pBABE-puro vector or a c-Myc2 expression vector. Cells were selected in 1 μg/ml puromycin for 2 more days and luciferase activity determined.
RNA Isolation and Northern Analysis.
Total RNA was isolated by using Trizol (GIBCO/BRL). Total RNA was then subjected to polyA+ selection with oligo dT cellulose (ICN). Approximately 2 μg polyA+ mRNA was separated on 1% agarose-5.4% formaldehyde denaturing gels and transferred. Blots were then UV crosslinked and prehybridized for 30 minutes with ExpressHyb (CLONTECH). Hybridization of probe was performed at 68°C for 1 hr with ExpressHyb. Probes were labeled with [α-32P]-dCTP (ICN) with the PrimeIt II random labeling kit (Stratagene). Blots were probed sequentially without stripping for p21CIP1 and cyclophilin. Northern blots for all HaCaT samples were probed with human p21CIP1 probe, whereas samples from NIH 3T3 cells were probed with murine p21CIP1 probe.
Western Blot Analysis.
Cells were lysed in RIPA buffer (1X PBS/1% Triton-X 100/0.5% sodium deoxycholate/0.1% SDS/30 μg/ml aprotinin/10 μg/ml PMSF) and sonicated briefly. Samples were normalized by standard protein assay (Bio-Rad), separated by SDS/PAGE and transferred to nitrocellulose (Schleicher & Schuell). Blots were probed overnight with the indicated primary antibodies and incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Jackson Laboratory) in 3% milk/TBS (20 mM Tris, pH 7.6/137 mM NaCl) buffer. Blots were washed with TBS.
Reporter Assays.
NIH 3T3 cells were plated at 5 × 105 cells/60-mm dishes and transfected 24 hr later, as described above. Cells were cotransfected with p21CIP1 luciferase vector, SV40-β-galactosidase (β-gal) and either empty CMV or CMV-Myc2. Cells were harvested 48 hr after transfection and lysed in Reporter Lysis Buffer (Promega). Luciferase activity was normalized by β-gal activity. Fold activity was then determined compared to empty vector control. Ectopic protein expression was confirmed by Western blot analysis to verify comparable expression (data not shown).
For stable NIH 3T3 p21CIP1 reporter lines, clonal lines were plated in 24-well plates. Cells were harvested 24 hr later and lysed in Promega lysis buffer. Luciferase activity was normalized by protein assay. For TSA tests, four clonal lines were seeded in 24-well plates and left untreated or treated with TSA at 500 ng/ml for 24 hr. Cells were then lysed in Reporter Lysis Buffer (Promega). Luciferase activity was normalized by standard protein assay (Bio-Rad). To determine the effects of TSA on p21CIP1 promoter activity, fold activity was determined by comparing luciferase activity of samples to that of untreated vector only control.
Results
Up-Regulation of p21CIP1 Correlates with Down-Regulation of c-Myc Protein After TGFβ Treatment of HaCaT Cells.
We sought to examine the effect of TGFβ activity on c-Myc and p21CIP1 expression. In separate studies, it has been shown that TGFβ treatment of epithelial cells leads to both up-regulation of p21CIP1 transcription and down-regulation of c-Myc (36–40). However, a temporal correlation between these events has not been demonstrated. To determine whether these events are related, we treated HaCaTs with TGFβ for the time periods indicated in Fig. 1. Western blot analysis revealed a rapid decrease in c-Myc proteins levels (Fig. 1A Upper). Down-regulation of c-Myc mRNA was confirmed by Northern blot analysis (data not shown). This down-regulation was paralleled by an up-regulation of p21CIP1 mRNA, as shown by Northern blot analysis (Fig. 1A Lower). Rapid increase of p21CIP1 mRNA expression occurred immediately after the rapid down-regulation of c-Myc protein, indicating that these events may be interdependent (Fig. 1B).
Figure 1.
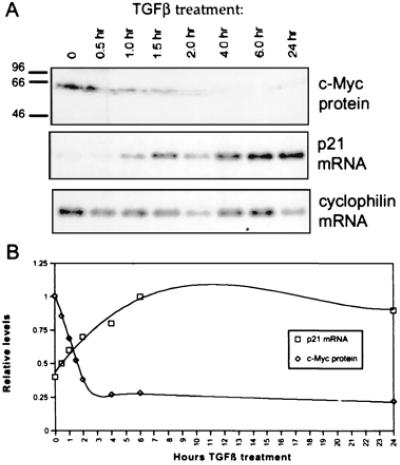
p21CIP1 up-regulation is temporally related to c-Myc down-regulation after TGFβ treatment of HaCaT cells. (A) HaCaT cells were treated with 1 ng/ml TGFβ for the time periods indicated. Samples were analyzed by Western blot with anti-Mycfl for c-Myc protein expression and by Northern blot for p21CIP1 mRNA expression. The Northern blot was subsequently probed for cyclophilin as a control for equal loading. (B) Densitometric analysis of Western and Northern blots. The highest value for each c-Myc protein and normalized p21CIP1 mRNA was set to 1.0 for graphical representation. c-Myc protein levels are indicated by diamonds, and p21CIP1 mRNA levels normalized to cyclophilin expression are represented by squares.
Inhibition of TGFβ Activity by c-Myc Is Associated with Repression of p21CIP1 mRNA Levels.
To understand further the relationship between c-Myc expression and TGFβ-mediated cell-cycle block, c-Myc was stably expressed in HaCaT cells by retroviral infection. Several monoclonal cell lines were isolated and analyzed for TGFβ responsiveness as well as c-Myc expression. Fig. 2A shows ectopically expressed c-Myc protein detected by an antibody specific for exogenous protein in two stable lines selected for analysis, Myc-cl1 (for Myc clone 1) and Myc-cl2. Treatment of vector-containing HaCaTs with TGFβ led to a decrease in levels of endogenous c-Myc, whereas HaCaTs ectopically expressing c-Myc exhibited no down-regulation of c-Myc protein (Fig. 2B). This stable expression of c-Myc protein resulted in an inhibition of TGFβ-mediated repression of DNA synthesis as determined by [3H]thymidine incorporation (Fig. 2C). This degree of inhibition of TGFβ activity by ectopic c-Myc expression is similar to that found in other cell lines (25). Therefore, enforced expression of c-Myc is capable of attenuating TGFβ activity in HaCaT cells.
Figure 2.
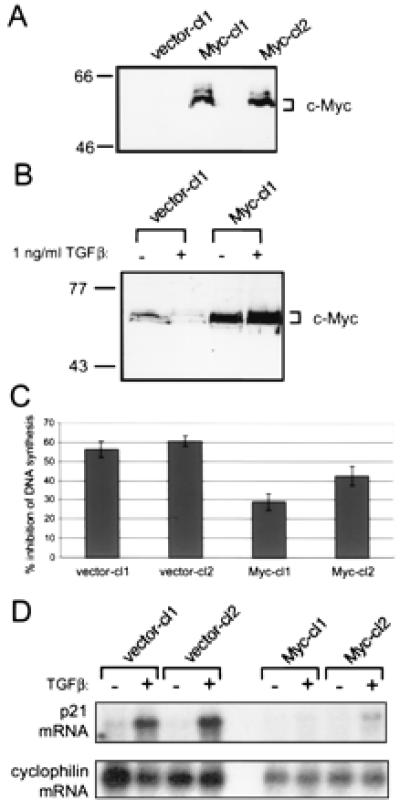
Expression of c-Myc protein in HaCaT lines blocks TGFβ-mediated cell-cycle arrest and subsequent up-regulation of p21CIP1 mRNA. (A) Ectopic expression of c-Myc in stable lines. Stable clonal HaCaT lines vector-cl1 (clone 1), Myc-cl1, and Myc-cl2 were analyzed for ectopic c-Myc expression by Western blot. Blots were probed with anti-av-myc 12C, which recognizes only exogenous c-Myc protein. (B) Down-regulation of c-Myc after TGFβ treatment. HaCaT clonal lines vector-cl1 and Myc-cl1 were left untreated or treated for 20 hr with 1 ng/ml TGFβ. Whole-cell lysates were analyzed by Western blot by using anti-Mycfl. (C) Inhibition of entry into S phase after TGFβ treatment. Cell-cycle inhibition by TGFβ treatment was determined by [3H]thymidine incorporation. Clonal HaCaT lines were plated in duplicate and re-fed the next day with fresh media with or without 1 ng/ml TGFβ. Cells were treated for 16 hr, then labelled with 1 mCi/ml [3H]thymidine for an additional 4 hr with or without TGFβ. [3H]thymidine incorporation was determined by scintillation counting. Inhibition of DNA synthesis is represented as the percent difference between untreated and treated cells. Error bars indicate standard deviation of the average of three independent experiments. (D) Up-regulation of p21CIP1 after TGFβ treatment is blocked by ectopic c-Myc expression. Clonal HaCaT control or c-Myc-expressing lines as indicated were left untreated or treated with 1 ng/ml TGFβ for 20 hr. PolyA+ mRNA was isolated, and 2 μg mRNA per sample was analyzed by Northern blot analysis. The blot was sequentially probed for both p21CIP1 (Top) and cyclophilin (Bottom).
Because c-Myc protein levels and p21CIP1 mRNA levels exhibited an inverse relationship after TGFβ treatment, we examined the expression of p21CIP1 in these clonal HaCaT lines stably expressing c-Myc. As shown in Fig. 2D, Northern blot analysis of vector-containing HaCaT lines demonstrated a dramatic up-regulation of p21CIP1 mRNA after TGFβ treatment. However, up-regulation of p21CIP1 message was effectively blocked in HaCaT lines ectopically expressing c-Myc (Fig. 2D). Thus, the block of TGFβ inhibition of cell-cycle progression may be because of repression of p21CIP1 transcription by c-Myc.
Overexpression of c-Myc Results in Repression of Basal Levels of p21CIP1 mRNA Levels.
Because up-regulation of p21CIP1 expression after TGFβ treatment was blocked by ectopic expression of c-Myc, we determined further whether overexpression of c-Myc could repress basal levels of p21CIP1 mRNA. NIH 3T3 cells were plated at high density (confluent at harvest) and lower density (subconfluent at harvest) and transfected with either vector only or c-Myc expression vector. Cells transfected with c-Myc exhibited high levels of c-Myc protein as compared to endogenous protein levels in vector-only transfected cells, as determined by Western blot analysis (data not shown). Northern blot analysis showed that p21CIP1 mRNA levels were unchanged in confluent cells (Fig. 3A) but diminished in subconfluent cells transfected with c-Myc (Fig. 3B). Densitometric analysis normalized to cyclophilin-loading control demonstrated that p21CIP1 levels were decreased approximately 50% in subconfluent c-Myc-transfected cells (Fig. 3 Bottom). Thus, overexpression of c-Myc can repress transcription of p21CIP1, although this effect depends on the density of the cells in culture.
Figure 3.
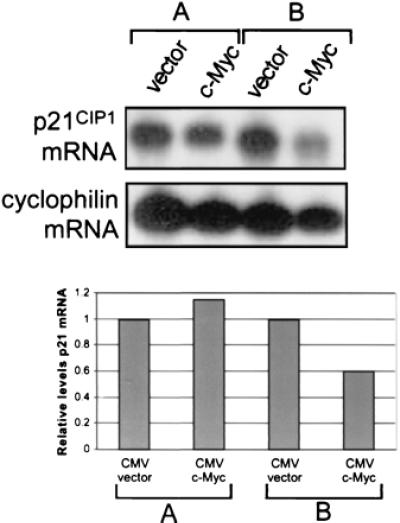
p21CIP1 levels are diminished in cells overexpressing c-Myc. NIH 3T3 cells were plated at 1.5 × 106 cells/100-mm dish (A) or 7 × 105 cells/100-mm dish (B) and transiently transfected 24 hr later with either empty CMV vector or CMV-Myc. PolyA+ mRNA was isolated, and approximately 1 μg RNA per sample was analyzed by Northern blot analysis. The blot was sequentially probed for both p21CIP1 (Top, Upper) and cyclophilin (Top, Lower). The Northern blot was then analyzed by densitometry, and relative levels of p21CIP1 mRNA normalized by levels of cyclophilin were plotted on a bar graph, where p21CIP1 levels in vector-only-transfected cells were set to a value of one for comparison with c-Myc transfected cells (Bottom).
Repression of p21CIP1 by c-Myc Occurs at the Promoter Level.
To determine whether repression of p21CIP1 transcription occurs at the promoter level, the role of c-Myc in p21CIP1 repression was analyzed further by reporter assay. NIH 3T3 cells were stably transfected with a 2.3-kb fragment of the p21CIP1 promoter linked to the luciferase reporter gene. Similar p21CIP1 reporter constructs have been used previously to identify major regulatory regions within this promoter (22, 23, 32, 41–43). After a short selection period, several clones were isolated and analyzed for luciferase activity. Four independent clones with varying levels of basal luciferase activity were subsequently infected with either empty vector or c-Myc expression vector. We found that c-Myc-repressed luciferase activity driven by the p21CIP1 promoter in all four lines by an average of 0.68-fold (Fig. 4A).
Figure 4.
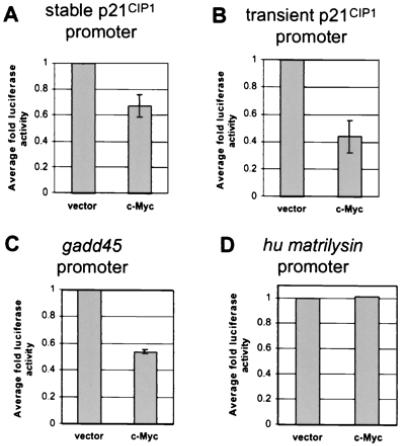
c-Myc-mediated transcriptional repression. (A) Repression of the p21CIP1 promoter in stable reporter lines by c-Myc. Generation of NIH 3T3 cell lines stably expressing p21CIP1-luciferase reporter constructs is described in Materials and Methods. Cells were analyzed for luciferase activity 24 hr after plating and normalized by standard protein assay. Fold luciferase activity was determined as compared to vector alone. Error bars represent standard deviation of the average of duplicate readings from four independent stable clones. (B) Repression of the p21CIP1 promoter by c-Myc. A 2.3-kb fragment of the p21CIP1 promoter linked to a luciferase reporter gene was transiently transfected into NIH 3T3 fibroblasts. Cells were cotransfected with a βgal vector for standardization and 1 μg of either empty CMV vector or c-Myc expression vector. Luciferase activity was determined 48 hr after transfection and normalized to β-gal activity. Fold luciferase activity was determined as compared to vector alone. Error bars represent standard deviation of the average of three independent experiments. (C) Repression of the gadd45 promoter by c-Myc. Reporter assays were performed as described for B, where 1 μg of either empty CMV or c-Myc expression vector was cotransfected into cells with a gadd45 luciferase reporter construct. Error bars represent standard deviation of the average of three independent experiments. (D) c-Myc expression does not affect transcription of the human matrilysin promoter. NIH 3T3 fibroblasts were transiently transfected with a reporter vector containing the human matrilysin promoter. Cells were then cotransfected with 1 μg of either empty CMV vector or c-Myc expression vector. Fold luciferase activity was determined as described for B.
To verify this repression by another method, we demonstrated further repression of the p21CIP1 promoter in NIH 3T3 cells by transient transfection. Transient transfection of c-Myc expression vector with this reporter construct in NIH 3T3 cells revealed that c-Myc repressed transcription from the p21CIP1 promoter by reporter assay an average of 0.44-fold (Fig. 4B). Transient transfection may result in slightly increased repression of the p21CIP1 promoter as compared to stable transfection, because expression levels of c-Myc are higher after transient transfection (data not shown). To compare the degree of repression of the p21CIP1 promoter by c-Myc to that of another c-Myc-repressed promoter, repression of the gadd45 promoter was analyzed. The fold repression of the gadd45 promoter in transient transfection assays (0.54 average fold) was found to be similar to that for the p21CIP1 promoter (Fig. 4C; compare to Fig. 4B). As a negative control, we examined a luciferase construct containing the human matrilysin promoter, which has not been shown to be regulated by c-Myc. The matrilysim promoter was not found to be affected by c-Myc expression (Fig. 4D). Similar results were found in transient transfections using a promoterless pGL2-Basic luciferase vector, demonstrating that c-Myc expression also does not affect basal luciferase expression or activity (data not shown). Therefore, the repression of the p21CIP1 promoter by c-Myc is specific.
Repression by c-Myc Does Not Involve Histone Deacetylase Activity.
One mechanism of transcriptional repression involves recruitment of histone deacetylase to a promoter. Deacetylation of histones is believed to prevent disassembly of nucleosomes, which results in DNA remaining inaccessible to transcriptional activation. To test this mechanism of repression, we treated four of the stable p21CIP1-reporter lines described above with TSA, an inhibitor of histone deacetylase activity. It has been demonstrated previously that TSA treatment activates transcription from the p21CIP1 promoter (42). TSA treatment of the p21CIP1 luciferase stable lines did result in up-regulation of luciferase activity in control lines containing empty retroviral vector only, where fold activity of TSA-treated cells was compared to untreated cells (Fig. 5A). Stable p21CIP1 reporter cell lines coinfected with either empty retroviral vector or c-Myc expression vector were then analyzed after TSA treatment. In this experiment, fold luciferase activity was calculated as compared to luciferase activity of untreated vector only cells (Fig. 5B). Although fold luciferase activity of TSA-treated c-Myc expressing cells was increased to approximately 1.8-fold, correspondingly the fold activity of vector-only control cells increased to 2.9-fold after TSA treatment. Thus, the relative fold activity of TSA-treated c-Myc expressing cells as compared to TSA-treated vector-only control cells was approximately 0.60-fold activity. This fold repression of luciferase activity of the p21CIP1 promoter is similar to that of untreated c-Myc expressing cells compared to vector-only controls (0.67-fold; Fig. 5B). Therefore, we conclude from this experiment that TSA treatment did not significantly affect repression of the p21CIP1 promoter by c-Myc, and that histone deacetylase activity is not required for Myc-mediated repression of p21CIP1.
Figure 5.
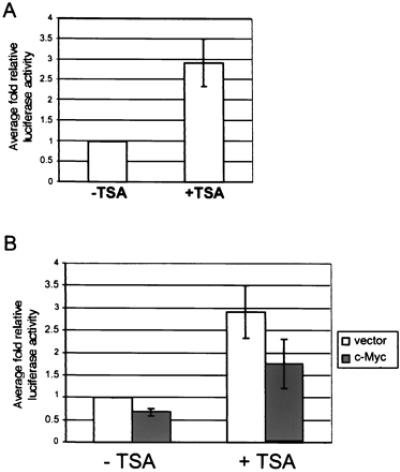
TSA treatment does not affect p21CIP1 repression by c-Myc. (A) Effect of TSA on basal p21CIP1 promoter activity. NIH 3T3 p21CIP1 reporter lines infected with vector only were left untreated (−TSA) or treated with 500 ng/ml TSA overnight (+TSA). Cells were analyzed for luciferase activity 24 hr after plating and normalized by standard protein assay. Fold luciferase activity was determined as compared to untreated cells for each line. Error bars represent standard deviation of the average of duplicate readings from four independent stable clones. (B) Effect of TSA on repression of the p21CIP1 promoter by c-Myc. NIH 3T3 p21CIP1 reporter lines infected with either empty retroviral vector (white bars) or c-Myc expression vector (dark bars) were left untreated (−TSA) or treated with 500 ng/ml TSA overnight (+TSA). Cells were analyzed for luciferase activity after treatment and normalized by standard protein assay. Fold luciferase activity was determined as compared to untreated vector only lines. Error bars represent standard deviation of the average of duplicate readings for four independent clones.
A Region Immediately Upstream of the Site of Transcriptional Initiation Is Sufficient for Repression of the p21CIP1 Promoter by c-Myc.
To analyze further c-Myc repression of the p21CIP1 promoter, we utilized 5′ deletions of the p21CIP1 promoter to identify the specific region of c-Myc regulation. Fig. 6A depicts binding sites for major regulatory proteins of p21CIP1 transcription as well as the TGFβ-responsive element, which has been identified as the region associated with up-regulation of p21CIP1 after TGFβ treatment (33). The specific deletion constructs are represented in Fig. 6B. As shown in the top diagram of Fig. 6B, the construct p21P Sma removes binding sites for p53 as well as C/EBP α and β. The deletion construct p21P Sma Δ1 further removes the TGFβ-responsive element (−86 to −71) and comes within approximately 20 nucleotides 5′ of the TATA box (black bar in diagrams). This element is then specifically removed in the deletion p21P SmaΔ2 (Fig. 6B Bottom). The full-length p21CIP1 promoter or the various deletion luciferase constructs were transiently transfected into cells and cotransfected with either empty CMV vector or CMV-Myc2. Repression of the p21CIP1 promoter by c-Myc was not significantly affected by deletion of p53 or C/EBP-binding sites (p21P Sma) (Fig. 6C). Further removal of the TGFβ-responsive element (p21P SmaΔ1) also did not affect repression by c-Myc. Finally, c-Myc-mediated repression of the p21CIP1 promoter was not affected by specific removal of the TGFβ-responsive element (p21P SmaΔ2). These results indicate that c-Myc repression does not overlap with the TGFβ-responsive element or the binding sites for p53 or C/EBP proteins, but appears to act through a region between −62 bp and +16 bp of the start site of transcription.
Figure 6.
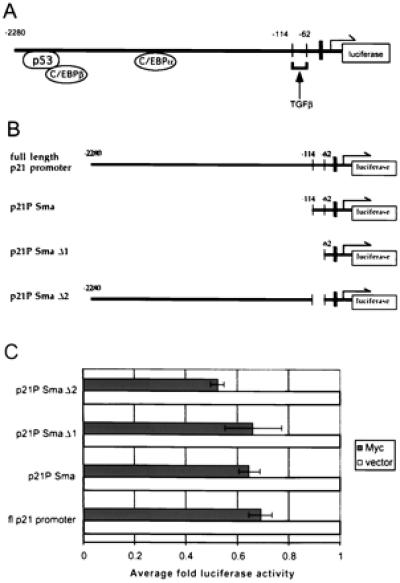
Sequences immediately upstream of the transcriptional initiation site are sufficient for repression of p21CIP1 by c-Myc. (A) Diagram of the p21CIP1 promoter illustrating regions bound by some of the major regulatory proteins, as well as the TGFβ-responsive element. The TATA box, located at −45 bp relative to start of transcription, is indicated by the black bar. The arrow indicates the site of transcriptional initiation. (B) Diagrams of 5′ deletion constructs of p21CIP1 promoter. Numbers in top three diagrams indicate 5′ end of the deletion construct relative to the transcription start site. The last diagram depicts deletion p21 pSmaΔ2, in which only the region from −114 to −62, including the TGFβ-responsive element, is deleted. (C) Luciferase activity for each promoter deletion construct depicted in B was determined, and fold luciferase activity in cells cotransfected with c-Myc expression vector (dark solid bars) was determined as compared to cells cotransfected with empty CMV vector (white solid bars). Error bars represent the standard deviation of the average of three independent experiments.
Discussion
In this paper, we have examined the role of Myc-mediated repression of p21CIP1 in TGFβ-mediated growth arrest. Enforced c-Myc expression has been shown previously to abrogate TGFβ-mediated cell cycle inhibition in murine keratinocytes (Balb/MK), murine myeloid leukemia cells (M1), and mink-lung epithelial cells (CCL64) (25–27). Here, we have extended these observations to include the immortalized HaCaT cell line. Furthermore, we demonstrated that c-Myc blocked up-regulation of p21CIP1 mRNA in TGFβ-treated cells. We also presented evidence that overexpression of c-Myc in subconfluent murine NIH 3T3 fibroblasts resulted in repressed levels of p21CIP1 mRNA. In support of these observations, we also have found that p21CIP1 mRNA levels are decreased in Myc/Ras cotransformed rat-embryo fibroblasts, as compared with cells transfected with Ras alone (unpublished observations). These results indicate further the ability of c-Myc to repress p21CIP1 transcription, because high levels of activated Ras have been shown to up-regulate p21CIP1 expression (44). Finally, we found that c-Myc repressed transcription of p21CIP1 at the promoter level. This repression did not depend on histone deacetylase activity and was not associated with previously identified regulatory elements, including the TGFβ-responsive element as well as binding sites for p53 and C/EBP α and β.
Functional Consequences of p21CIP1 Repression by c-Myc.
c-Myc activity affects many cellular functions, including cell proliferation, apoptosis, and immortalization. We have demonstrated here that repression of p21CIP1 is linked to c-Myc's ability to promote cell-cycle progression. Indeed, in terms of TGFβ-dependent cell-cycle inhibition, immediate down-regulation of c-Myc and induction of p21CIP1 have been shown independently to be important events (40). It has also been shown that inactivation of c-Myc activity by expression of a mutant Max protein led to G0/G1 phase extension coupled with increases in p21CIP1 expression (45). Finally, overexpression of c-Myc in TPA-sensitive cancer cells blocked cell-cycle inhibition and was associated with repression of p21CIP1 expression (46). Coinfection of these cancer cells with both c-Myc and p21CIP1 resulted in cell-cycle arrest, indicating that suppression of p21CIP1 is an important event in Myc-dependent cell-cycle progression. Our results demonstrating that repression of p21CIP1 transcription by c-Myc is associated with cell-cycle promotion provide further insight into the pathways by which c-Myc regulates cell-cycle progression.
p21CIP1 expression is highly regulated by a variety of factors that either activate or repress transcription. This regulation of p21CIP1 levels in the cell is critical for the functional effects of p21CIP1 expression, because it has been shown that at low levels, p21CIP1 may serve as a scaffold for active cyclin/cdk complexes, whereas higher levels of p21CIP1 inhibit cyclin/cdk activity (47–49). Thus, the balance of factors regulating p21CIP1 transcription may determine the role of p21CIP1 expression in the cell. Deregulated c-Myc expression in cancer may be critical in tipping the balance in favor of repression of p21CIP1 transcription. Although exogenous overexpression of c-Myc may not reflect a true physiological situation, deregulated expression of c-Myc in cancer represents a loss of normal regulation. Thus, examination of p21CIP1 transcription in cells engineered to overexpress c-Myc is useful as a model to recapitulate deregulated expression of c-Myc in cancer. To gain some insight into regulation of p21CIP1 levels in the absence and presence of c-Myc, we have examined quiescent and serum-restimulated HaCaT cells and human foreskin fibroblasts. An inverse relationship exists between c-Myc and p21CIP1 expression as cells become growth inhibited after serum deprivation and during subsequent restimulation (unpublished observations).
The recent development of c-Myc-null Rat1 fibroblasts provides a system in which to examine gene expression in the absence of c-Myc (50). Analysis of expression of putative c-Myc-regulated genes in this cell line revealed that several genes previously reported to be activated by c-Myc do not exhibit significant alterations in expression, with the exception of cad (51). However, autorepression of the c-Myc promoter as well as repression of gadd45 was shown to be altered in the c-Myc null fibroblasts as compared to either the wild-type c-Myc parental Rat1 cell line or cells in which c-Myc expression was restored (51). We attempted to analyze p21CIP1 expression in these cells; however, we were unable to detect p21CIP1 (unpublished observations). Similarly, other groups have been unable to detect p21CIP1 in Rat1 fibroblasts (52–53). Although it was found that p21CIP1 protein levels appear to be diminished in the c-Myc-null Rat1 cells (54), other recent reports support repression of p21CIP1 by c-Myc (46, 55).
Repression of p21CIP1 at the Promoter Level.
In this report, we demonstrate that c-Myc transcriptionally represses the p21CIP1 promoter. Furthermore, we localized the region of this repression within approximately 60 nucleotides upstream of the start site of transcription. Any further deletions would most likely impact on basal transcription, because the TATA box is located at −47 bases relative to the transcription start site. This region does not overlap with previously identified regions of regulation by factors such as p53 or C/EBP proteins (56). The finding that HaCaT cells have two mutant alleles for p53 (39) supports our data, demonstrating that repression of the p21CIP1 promoter is independent of the p53-binding site. Finally, we demonstrated that deletion of the region associated with up-regulation after TGFβ signaling also does not affect repression of this promoter by c-Myc. Therefore, c-Myc repression of the p21CIP1 promoter involves a mechanism independent of these regulatory regions.
Because c-Myc is capable of repression of the p21CIP1 promoter through a region immediately upstream of the transcriptional start site, we tested whether repression involved histone deacetylase activity. Histone deacetylase is recruited to promoters by many transcriptional repressors, including Mad proteins, which compete with c-Myc for binding to Max (57–59). Also, it has been shown that transcription from the p21CIP1 promoter is activated after treatment with TSA, a histone deacetylase inhibitor (42). We utilized cell lines stably transfected with a p21CIP1 promoter reporter vector, because chromatin assembly may not be complete in transient transfections. Our results indicate that c-Myc represses p21CIP1 transcription by a mechanism independent of histone deacetylase activity.
With the exception of C/EBP-binding sites, the p21CIP1 promoter lacks other known Myc-binding sequences, including E-box Myc sequences (EMS) and Inr elements (60, 61). Similarly, the gadd45 promoter, which is also repressed by c-Myc, also lacks EMS or Inr sequences. However, repression of these genes by c-Myc may not involve direct DNA binding. c-Myc may repress gene transcription by binding directly to transcriptional components and preventing formation of an active preinitiation complex. Alternatively, c-Myc may bind the preinitiation complex and prevent transcriptional initiation. Indeed, it has been shown that c-Myc binds TATA box-binding protein, a key component of the basal transcriptional machinery (62, 63). Further investigation is necessary to clarify the mechanisms by which c-Myc mediates gene repression.
Acknowledgments
We thank Scott Lowe for providing the murine ecotropic receptor virus construct, Xiao-Fan Wang for the deletion constructs p21pSma, p21 pSmaΔ1, and p21 pSmaΔ2, Wafik El-Deiry for a Northern probe for murine p21CIP1, and Rebecca Chinery for numerous reagents, as well as invaluable discussions. We also thank Howard Crawford for helpful discussions, and Linda Penn and Mark Gregory for critical reading of the manuscript. This work was supported by Public Health Service Grant CA47399 from the National Cancer Institute.
Abbreviations
- TGFβ
transforming growth factor β
- TSA
trichostatin A
- HaCaT
human immortalized keratinocytes
- β-gal
β-galactosidase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.150006697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.150006697
References
- 1.Mateyak M K, Obaya A J, Adachi S, Sedivy J M. Cell Growth Diff. 1997;8:1039–1048. [PubMed] [Google Scholar]
- 2.Karn J, Watson J V, Lowe A D, Green S M, Vedeckis W. Oncogene. 1989;4:773–787. [PubMed] [Google Scholar]
- 3.Eilers M, Schirm S, Bishop J M. EMBO J. 1991;10:133–141. doi: 10.1002/j.1460-2075.1991.tb07929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole M D, McMahon S B. Oncogene. 1999;18:2916–2924. doi: 10.1038/sj.onc.1202748. [DOI] [PubMed] [Google Scholar]
- 5.Claassen G F, Hann S R. Oncogene. 1999;18:2925–2933. doi: 10.1038/sj.onc.1202747. [DOI] [PubMed] [Google Scholar]
- 6.Obaya A J, Mateyak M K, Sedivy J M. Oncogene. 1999;18:2934–2941. doi: 10.1038/sj.onc.1202749. [DOI] [PubMed] [Google Scholar]
- 7.Dang C V. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Facchini L M, Penn L Z. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- 9.Grandori C, Eisenman R N. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- 10.Henrikkson M, Luscher B. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- 11.Massague J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 12.Hoodless P A, Wrana J L. Curr Top Microbiol Immunol. 1998;228:235–272. doi: 10.1007/978-3-642-80481-6_10. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrow M G, Moses H L. Cancer Res. 1995;55:1452–1457. [PubMed] [Google Scholar]
- 14.Ravitz M J, Wenner C E. Adv Cancer Res. 1997;71:165–207. doi: 10.1016/s0065-230x(08)60099-8. [DOI] [PubMed] [Google Scholar]
- 15.Alevizopoulos A, Mermod N. BioEssays. 1997;19:581–591. doi: 10.1002/bies.950190709. [DOI] [PubMed] [Google Scholar]
- 16.Warner B J, Blain S W, Seoane J, Massague J. Mol Cell Biol. 1999;19:5913–5922. doi: 10.1128/mcb.19.9.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr C J, Roberts J M. Genes Dev. 1999;15:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 18.Johnson D G, Walker C L. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- 19.Noda A, Ning Yi, Venable S F, Pereira-Smith O M, Smith J R. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 20.El-Deiry W, et al. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 21.Timchenko N A, Wilde M, Nakanishi M, Smith J R, Darlington G J. Genes Dev. 1996;10:804–815. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 22.Chinery R, Brockman J A, Peeler M O, Shyr Y, Beauchamp R D, Coffey R J. Nat Med. 1997;3:1233–1241. doi: 10.1038/nm1197-1233. [DOI] [PubMed] [Google Scholar]
- 23.Datto M B, Hu P P-C, Kowalik T F, Yingling J, Wang X-F. Mol Cell Biol. 1997;17:2030–2037. doi: 10.1128/mcb.17.4.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michieli P, Chedid M, Lin D, Pierce J H, Mercer W E, Givol D. Cancer Res. 1994;54:3391–3395. [PubMed] [Google Scholar]
- 25.Alexandrow M G, Kawabata M, Aarke M, Moses H L. Proc Natl Acad Sci USA. 1995;92:3239–3243. doi: 10.1073/pnas.92.8.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selvakumaran M, Lin H K, Sjin R T, Reed J C, Liebermann D A, Hoffman B. Mol Cell Biol. 1994;14:2352–2360. doi: 10.1128/mcb.14.4.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longstreet M, Miller B, Howe P H. Oncogene. 1992;7:1549–1556. [PubMed] [Google Scholar]
- 28.Spotts G D, Patel S V, Xiao Q, Hann S R. Mol Cell Biol. 1997;17:1459–1468. doi: 10.1128/mcb.17.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hann S R, Abrams H D, Rohrschneider L R, Eisenman R N. Cell. 1983;34:789–798. doi: 10.1016/0092-8674(83)90535-4. [DOI] [PubMed] [Google Scholar]
- 30.Hann S R, Dixit M, Sears R C, Sealy L. Genes Dev. 1994;8:2441–2452. doi: 10.1101/gad.8.20.2441. [DOI] [PubMed] [Google Scholar]
- 31.Spotts G D, Hann S R. Mol Cell Biol. 1990;10:3952–3964. doi: 10.1128/mcb.10.8.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Deiry W S, et al. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- 33.Datto M B, Yu Y, Wang X-F. J Biol Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- 34.Marhin W W, Chen S, Facchini L M, Fornace A J, Penn L Z. Oncogene. 1997;14:2825–2834. doi: 10.1038/sj.onc.1201138. [DOI] [PubMed] [Google Scholar]
- 35.Gaire M, Magbunua Z, McDonnell S, McNeil L, Lovett D H, Matrisian L M. J Biol Chem. 1994;269:2032–2040. [PubMed] [Google Scholar]
- 36.Coffey R J, Bascom C C, Sipes N J, Graves-Deal R, Weissman B E, Moses H L. Mol Cell Biol. 1988;8:3088–3093. doi: 10.1128/mcb.8.8.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietenpol J A, Holt J T, Stein R W, Moses H L. Proc Natl Acad Sci USA. 1990;87:3758–3762. doi: 10.1073/pnas.87.10.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landesman Y, Pagano M, Draetta G, Rotter V, Fusenig N E, Kimchi A. Oncogene. 1992;7:1661–1665. [PubMed] [Google Scholar]
- 39.Datto M B, Li Y, Panus J F, Howe D J, Xiong Y, Wang X-F. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malliri A, Yeudall W A, Nikolic M, Crouch D H, Parkinson E K, Ozanne B. Cell Growth Diff. 1996;7:1291–1304. [PubMed] [Google Scholar]
- 41.El-Deiry W S, Tokino T, Velculecu V E, et al. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 42.Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, Sakai T. Biochem Biophys Res Commun. 1997;241:142–150. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- 43.Billon N, van Grunsven L A, Rudkin B B. Oncogene. 1996;13:2047–2054. [PubMed] [Google Scholar]
- 44.Kerkhoff E, Rapp U R. Oncogene. 1998;17:1457–1462. doi: 10.1038/sj.onc.1202185. [DOI] [PubMed] [Google Scholar]
- 45.Borre A, Cultrarto C M, Segal S. J Cell Phys. 1996;169:200–208. doi: 10.1002/(SICI)1097-4652(199610)169:1<200::AID-JCP20>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell K O, El-Deiry W S. Cell Growth Diff. 1999;10:223–230. [PubMed] [Google Scholar]
- 47.Zhang H, Hannon G J, Beach D. Genes Dev. 1994;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Hannon G J, Casso D, Beach D. Cold Spring Harbor Symp Quant Biol. 1994;59:21–29. doi: 10.1101/sqb.1994.059.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Harper J W, Elledge S J, Keyomarsi K, Dynlacht B, Tsai L H, Zhang P, Dobrowolski S, Bai C, Connell C L, Swindell E, et al. Mol Cell Biol. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mateyak M K, Obaya A J, Adachi S, Sedivy J M. Cell Growth Diff. 1997;8:1039–1048. [PubMed] [Google Scholar]
- 51.Bush A, Mateyak M, Dugan K, et al. Genes Dev. 1998;12:3797–3802. doi: 10.1101/gad.12.24.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steiner P, Philipp A, Lukas J, Godden-Kent D, Pagano M, Mittnacht S, Bartek J, Eilers M. EMBO J. 1995;14:4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez-Roger I, Solomon D L C, Sewing A, Land H. Oncogene. 1997;14:2373–2381. doi: 10.1038/sj.onc.1201197. [DOI] [PubMed] [Google Scholar]
- 54.Mateyak M K, Obaya A J, Sedivy J M. Mol Cell Biol. 1999;19:4672–4683. doi: 10.1128/mcb.19.7.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coller H A, Grandori C, Tamayo P, Colbert T, Lander E S, Eisenman R N, Golub T R. Proc Natl Acad Sci USA. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gartel A L, Tyner A L. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 57.Heinzel T, Lavinsky R M, Mullen T M, et al. Nature (London) 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 58.Alland L, Muhle R, Hou H, Jr, Potes J, Chin L, Schreiber-Agus N, DePinho R A. Nature (London) 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 59.Nagy L, Kao H Y, Chakravarti D, Lin R J, Hassig C A, Ayer D E, Schreiber S L, Evans R M. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 60.Li L-h, Nerlov C, Prendergast G, MacGregor D, Ziff E B. EMBO J. 1994;13:4070–4079. doi: 10.1002/j.1460-2075.1994.tb06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roy A L, Carruthers C, Gutjahr T, Roeder R G. Nature (London) 1993;365:359–361. doi: 10.1038/365359a0. [DOI] [PubMed] [Google Scholar]
- 62.Hateboer G H, Timmers H, Rustgi A, Billaud M, van't Veer L, Bernards R. Proc Natl Acad Sci USA. 1993;90:8489–8493. doi: 10.1073/pnas.90.18.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maheswaran S, Lee H, Soneneshein G E. Mol Cell Biol. 1994;14:1147–1152. doi: 10.1128/mcb.14.2.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]