

Abstract
Several lines of evidence suggest that the serotonin (5-hydroxytryptamine, 5-HT) regulates cardiovascular functions during embryogenesis and adulthood. 5-HT binds to numerous cognate receptors to initiate its biological effects. However, none of the 5-HT receptor disruptions in mice have yet resulted in embryonic defects. Here we show that 5-HT2B receptor is an important regulator of cardiac development. We found that inactivation of 5-HT2B gene leads to embryonic and neonatal death caused by heart defects. 5-HT2B mutant embryos exhibit a lack of trabeculae in the heart and a specific reduction in the expression levels of a tyrosine kinase receptor, ErbB-2, leading to midgestation lethality. These in vivo data suggest that the Gq-coupled receptor 5-HT2B uses the signaling pathway of tyrosine kinase receptor ErbB-2 for cardiac differentiation. All surviving newborn mice display a severe ventricular hypoplasia caused by impaired proliferative capacity of myocytes. In adult mutant mice, cardiac histopathological changes including myocyte disarray and ventricular dilation were consistently observed. Our results constitute genetic evidence that 5-HT via 5-HT2B receptor regulates differentiation and proliferation of developing and adult heart. This mutation provides a genetic model for cardiopathy and should facilitate studies of both the pathogenesis and therapy of cardiac disorders in humans.
Keywords: neuregulin, knockout, proliferation, transactivation
Serotonin (5-hydroxytryptamine) (5-HT) first isolated as a vasoconstrictor from blood was later identified in the central nervous system (CNS). It is found in three main areas of the body: the intestinal wall, platelets, and CNS. The functions of 5-HT in CNS as a neurotransmitter are numerous and appear to involve control of appetite, sleep, memory and learning, temperature regulation, mood, behavior (including sexual and hallucinogenic behavior), and endocrine regulation (1). Peripherally, 5-HT, which is stored in platelets, appears to play a major role in homeostasis, blood pressure regulation, cardiovascular functions (2), motility of the gastrointestinal tract (3), and carcinoid tumor pathology (4). Independently of its location in adults, 5-HT also has been detected during zygotic cleavage divisions, gastrulation and neurulation in embryos of sea urchins, frogs, chicken, and Drosophila. The presence of 5-HT and its receptors in early embryogenesis and the ability of 5-HT-specific pharmacological agents (5) to interfere with embryonic development suggested that early embryos use 5-HT before the onset of neurogenesis to regulate cell proliferation and/or morphogenetic movements (6, 7). Furthermore, 5-HT has been suspected for years to regulate craniofacial and cardiovascular morphogenesis: In embryos grown in the presence of either a high concentration of 5-HT or 5-HT-specific reuptake inhibitors, a decreased proliferation of myocardium, cardiac mesenchyme, and endothelium has been reported, indicating that 5-HT may regulate proliferation in the embryonic heart (8).
These biological actions of 5-HT are mediated by numerous cognate receptors. It now appears that there are at least 15 receptor subtypes that belong to four populations: 5-HT1/5, 5-HT2, 5-HT3, and 5-HT4/6/7 subtypes (9). This diversity has limited the use of a pharmacological approach toward the understanding of the specificity of these various receptors. An alternative way of investigation is genetic inactivation of genes encoding these receptors. Until now, the phenotype obtained by homologous recombination of 5-HT receptor genes (5-HT1A, 5-HT1B, or 5-HT2C) has led exclusively to behavioral abnormalities without reported morphological defects in CNS (10). The receptors required for the peripheral 5-HT functions still need to be identified.
The 5-HT2 receptor family comprises three subtypes: 5-HT2A, 5-HT2B, and 5-HT2C. The 5-HT2B receptor belongs to the G protein-coupled seven transmembrane-spanning receptor family. The 5-HT2B receptor is expressed in embryonic (11) and adult (12) cardiovascular tissues, gut and brain from the rat, mouse, and human species. Binding of 5-HT to this receptor activates Gαq/11 protein, thereby activating phospholipase C, which initiates a rapid release of inositol trisphosphate, resulting in an increase in intracellular calcium levels in transfected cells. Recently, this receptor has been shown to activate the phospholipase A2 (13) and nitric oxide synthases (14) in transfected and endogenously 5-HT2B-expressing cells. Agonist stimulation of 5-HT2B receptor subtype also causes a rapid and transient activation of the protooncogene product p21ras (15) and p42mapk/p44mapk mitogen-activated protein kinases and transduces a mitogenic signal by activating the nonreceptor tyrosine kinase c-Src and the receptor tyrosine kinase platelet-derived growth factor receptor (16) in mouse fibroblast cells. 5-HT2B receptor expression has been shown to induce tumors in nude mice and has been detected at a high level in human carcinoid tumors (15). By using a mouse embryo culture technique, treatment with broad spectrum 5-HT2 receptor antagonists resulted in severe morphological defects of neuroectodermal and mesodermal derivatives including ventricular myocardium (11). These differentiation processes may be controlled, at least in part, by the mitogenic properties of 5-HT2B receptor and integration of receptor tyrosine kinase-signaling pathways. However, the molecular mechanism by which 5-HT regulates embryonic development is currently unknown because none of the inactivated 5-HT receptors has yet been reported to induce obvious developmental defects other than behavioral defects (10).
Here it is shown that targeted inactivation of the 5-HT2B receptor gene leads to disturbed heart development, thereby resulting in embryonic lethality and death in neonatal mice. Our results identify a 5-HT2B receptor-mediated pathway by which 5-HT interacts with ErbB-2 to control differentiation in developing heart. Together, these results reveal 5-HT via 5-HT2B receptor as an important regulator of cardiac myocyte differentiation and proliferation.
Materials and Methods
5-HT2B Receptor Gene Targeting and Genotype Analysis.
The targeting vector was constructed with the selective bacterial neo gene introduced into the exon 2 sequence of the 5-HT2B receptor locus. This resulted in a nonfunctional allele of 5-HT2B because it interrupts the protein reading frame. One recombination event was obtained after screening of ≈4,000 embryonic stem (ES) cells clones. Southern blot analysis on the recombined clone with a probe derived from the neo sequence revealed that it contained a single expected 16-kbp fragment in a HpaI restriction enzyme digestion excluding multiple insertion of the targeting vector (not shown). From this recombined 5-HT2B ES cells strain, 13 chimeric animals were created by injection into C57/BL6 blastocyst embryos. These chimeras were bred to either the C57/BL6 or 129/PAS mouse strain to obtain germ-line transmission. After transfection of the 5-HT2B targeting vector into 129/PAS ES cells, recombined clones were analyzed by Southern blot with a probe (P) located 3′ to the sequence present in the targeting vector. Tail DNAs from newborn mice derived from intercrosses between heterozygous mice were digested with BglII and analyzed by Southern blot with probe P. All data have been obtained with mice bearing the mutation in a pure 129/PAS background, but similar defects have been observed in a mixed C57/BL6–129/PAS background.
Analysis of Mouse 5-HT2 Receptor Expression.
Membrane proteins prepared from 6-week-old mice heart ventricles (n = 4) or stomach (n = 5) were analyzed by binding studies with specific tritiated antagonists of the 5-HT2B ([3H]LY266070), 5-HT2A ([3H]MDL100907), or 5-HT2C ([3H]Mesulergine) receptors as described (11).
Morphological Analysis of Mouse Embryos.
Electron microscopy and histological experiments were performed as described (11). The hearts were fixed in paraformaldehyde, dehydrated and paraffin-embedded, then sectioned (7 μm) transversely from the apex, and sections were examined after Masson's trichrome staining. The hearts immersed in a cardioplegic solution (25 mM KCl/5% dextrose in PBS) to ensure complete myocardial relaxation before the hearts were fixed for transmission electron microscopy (TEM). Immunocytochemical labeling of embryos with a 5-HT2B-specific antibody on cryosections or anti-ErbB-2 (SC-284)-specific antibody (Santa Cruz Biotechnology) on paraffin sections was performed as described (11). Whole-mount immunohistochemical staining of embryos for developing blood vessels with antiplatelet endothelial cell adhesion molecule (PharMingen 01951D) antibody were performed as described (11). Western blot analysis for ErbB-2 and platelet-derived growth factor receptor-β (SC-432) was performed in the whole embryo or cell lysate as described (16).
Analysis of ErbB-2 mRNA Expression.
Semiquantitative reverse transcription–PCR was performed on 1 μg of total RNA for 30 cycles at an annealing temperature of 55°C. Normalization was performed by using RNA for elongation factor 1a. The oligonucleotides used for ErbB-2 are 5′-cgtgctagtcaagagtcccaacc and 5′-tactcttcagcatcgaccagctc; for elongation factor 1a, 5′-acaaactgaaagctgagcgtg and 5′-gtgcatttccacagacttgac; and for ErbB-4, 5′-tgaacaatgtgatggcaggtgc and 5′-cacgaagttatgaggacatttc.
Analysis of Proliferation Rate.
Ventricular cardiomyocytes were prepared from the hearts of newborn and 9.5-day postcoitum (dpc) embryos from 5-HT2B mutant and wild-type mice according to Adams et al. (17). The hearts were excised and kept in the buffer containing 113 mM NaCl/4.7 mM KCl/0.6 mM NaH2PO4/0.6 mM Na2HPO4/1.2 mM MgSO4/12 mM NaHCO3/20 mM glucose/10 mM Hepes. The atrium was removed and the ventricle was minced and then cells were dispersed by gentle agitation or gentle pipetting (for embryos). After 15 min of enzymatic digestion with collagenase type II and pancreatin at 37°C, myocytes were pelleted and washed, and differentially plated for 1 h to eliminate remaining nonmyocytes. Cells then were briefly pelleted at low speed (800 g) and rinsed twice in buffer. Rod-shaped cardiac myocytes were plated in culture medium on gelatin (2%)-precoated 8-well chamber slides in the presence of antimitotic (cytosine arabinoside) overnight. Cardiomyocytes were kept in DMEM with 10% FCS and antibiotics at a density of 103 cells per well. Approximately 95% of the cells displayed spontaneously contractile activity in culture and were verified by myosin heavy chain staining.
Thymidine incorporation was detected in cardiomyocytes as described (16). Briefly, quiescent cells (24-h serum starved) were treated with 5-HT (1 μM) or neuregulin (heregulin) (25 μg/ml) for 16 h, and 0.5 mCi of [3H]thymidine was added to the culture during the last 4 h of incubation. The free radioactivity was washed in 5% trichloroacetic acid, and the incorporated radioactive thymidine was quantified by scintillation counting. BrdUrd incorporation was performed by using a BrdUrd-labeling and staining kit (Boehringer Mannheim). Newborn mice were i.p. injected with BrdUrd; 4 h after injection, hearts were dissected and cardiac cryosections were stained with BrdUrd antibody. The sections were washed with water and mounted in 4′,6-diamidino-2-phenylindole-containing medium. Slides were kept at 4°C in the dark.
Results and Discussion
To generate 5-HT2B mutant mice, we produced ES cells in which one 5-HT2B allele was disrupted by homologous recombination (Fig. 1a). After injection into the blastocyst of the recombined ES cells, we obtained chimeras that transmitted the mutation. The genotype of 5-HT2B recombinant mice was determined by Southern blot analysis of tail DNA (Fig. 1b). The lack of functional 5-HT2B receptors was confirmed at the RNA level by reverse transcription–PCR amplification (data not shown) and the protein level by binding experiments with receptor subtype-specific-labeled antagonists. In addition to a loss of 5-HT2B receptor expression, no compensatory overexpression of either 5-HT2A or 5-HT2C receptors in heart and stomach tissues from 5-HT2B homozygous mice was detected (Fig. 1c). Heterozygous 5-HT2B mutant intercrosses resulted in a frequency of homozygous pups in newborns of only 16.7%, significantly different from the expected 25% (P < 0.05, n = 120, according to an unpaired t test), suggesting some embryonic lethality (Fig. 1d). When embryos were examined, a Mendelian ratio of homozygous mutant was seen up to 10.5 dpc. The surviving curve for mutants shows the periods of lethality before 11.5 dpc and during the first week of life (Fig. 1e). Nevertheless, the growth of some mutant embryos was clearly retarded at 9.5 and 10.5 dpc, exhibiting enlarged hearts and a pooling of blood in the abdominal area. Closer examination revealed that this was caused by blood leakage into the pericardial cavity (Fig. 2a). Surviving homozygous embryos at 12.5 dpc exhibited developmental delays and paler color than wild-type littermates (Fig. 2b). Because the 5-HT2B receptor is expressed in embryonic (11) and adult (12) cardiovascular tissues, including myocardial, endothelial, and vascular smooth muscle cells, we reasoned that defects observed in the mutant embryos might reflect problems in either vascular organization or cardiac morphogenesis. Whole-mount immunohistochemical staining of 10.5 dpc mutant embryos by using a mAb against platelet endothelial cell adhesion molecule as a marker of developing blood vessels revealed no gross defects in vascular patterning (Fig. 2c). Histological analysis of hearts from 5-HT2B mutant embryos revealed defects in the subepicardial layer, and a lack of trabecular cells in the ventricle (Fig. 2 d and e). However, the endocardial cushion appeared to be normal or only slightly reduced in size in some mutant embryos. The atrium and the outflow tract of the mutants appeared normal. The myocardial abnormalities in the 5-HT2B mutants correlate with the pattern of 5-HT2B receptor expression in the compact zone and the myocardial trabeculae (Fig. 3c). TEM studies revealed that all observed mutant hearts (n = 6) at 9.5 dpc display premature differentiation of sarcomeres (the contractile unit found in differentiated myocardial cells) within the compact zone (Fig. 2f), even within regions of the heart showing otherwise nearly normal trabeculation. The most severe reduction in the thickness of the myocardium is likely to induce myocardial rupture, resulting in escape of blood into the pericardium and death. Because we reported that 5-HT2B receptor transduces mitogenic signals in fibroblasts (15, 16), impaired differentiation, migration, and/or proliferation in the heart of 5-HT2B-mutant embryos probably accounts for the midgestation lethality. 5-HT developmental action has been suspected to trigger embryopathies including cardiac pathology observed in embryos from phenylketonuria patients who exhibit a low level of 5-HT in their blood (18).
Figure 1.
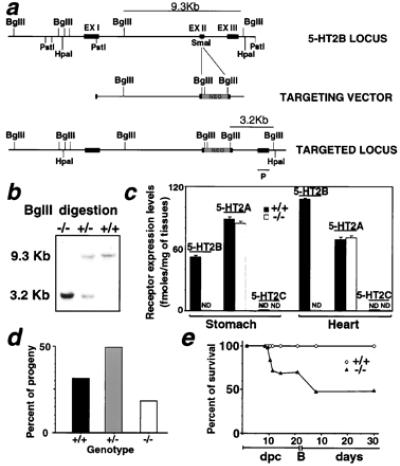
Targeted disruption of the 5-HT2B receptor gene. (a) (Top) The restriction map of the 5-HT2B receptor genomic locus of interest; (Middle) the targeting construct; and (Bottom) the mutated locus after homologous recombination. (b) The wild-type (+/+), heterozygous (+/−), and homozygous mutant (−/−) alleles were detected by Southern blot analysis. The probe (P) located outside of the construct detects a 9.3-kbp BglII fragment from wild-type DNA whereas it is reduced to 3.2-kbp in the homologous recombined allele. (c) The analysis of receptor membrane proteins by binding assay revealed a complete loss of 5-HT2B-specific sites in stomach and heart tissues of mutant mice. The maximal amount of binding sites is expressed as fmol specific binding/mg tissues ± SEM (n > 4). ND, no specific binding detected. (d) Genotyped progeny from 5HT2B +/− intercrosses showed that the homozygous genotype did not exhibit a Mendelian ratio, indicating some embryonic lethality among the homozygous. The percent of progeny is calculated from 120 newborn mice from 14 different litters. The homozygous mutant (−/−) (22 out of 120) represented 66% of the expected viable calculated, assuming sum of +/+ and +/− is 75%. (e) Percentage of homozygous survival before and after the birth in the progeny of heterozygous crosses. Before birth (B), the percent of survival is estimated from more than four litters of heterozygous crosses (n > 30), and a Mendelian ratio is observed until 10.5 dpc.
Figure 2.
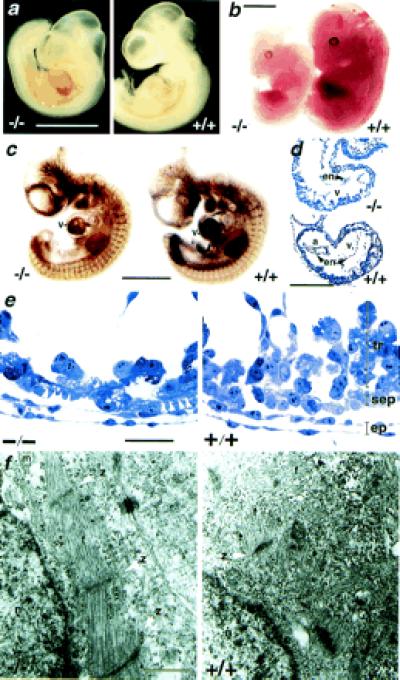
Morphology of 5-HT2B mutant embryos. (a) 5-HT2B-deficient embryos at 10.5 dpc exhibit a typical bleeding into the pericardial cavity. (b) At 12.5 dpc, the mutant embryos are smaller and paler than their wild-type littermates. (c) Whole-mount immunohistochemical staining of 10.5 dpc embryos for developing blood vessels with platelet endothelial cell adhesion molecule antibody revealed no gross defects in vascular pattering compared with age-matched wild-type littermates but reduced staining in heart ventricle (v). (d) Semithin sagittal sections of 9.5 dpc embryos demonstrate a severe reduction in the thickness of the ventricle (v) including both the compact zone and the trabeculae in 5-HT2B-deficient embryos. a, Atrium; en, endocardium. (e) Higher magnification shows a reduction of trabecular cells (tr) in the mutant heart, whereas elongated cells (white arrowhead) are visible in the compact subepicardial layer (sep). Epicardial cells (ep) are normally developed. (f) TEM analysis of these embryos reveals the abnormal sarcomeric differentiation within the subepicardial layer in all observed mutant heart (n = 6) but not in wild-type heart. z, Z band of the sarcomeres; f, actin fibers; m, mitochondria; and n, nucleus. Genotype designations are +/+, wild type; and −/−, homozygous mutant. (Bars for a–c = 500 μm; d = 100 μm; e = 5 μm, and f = 0.5 μm.)
Figure 3.
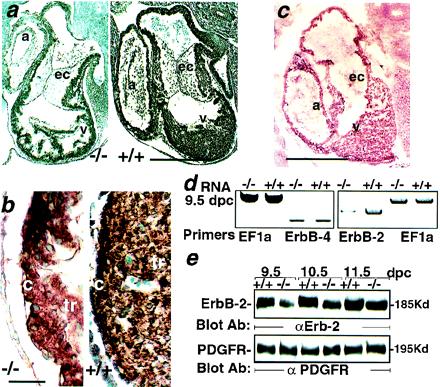
Reduced ErbB-2 expression in 5-HT2B mutant embryos. (a) The immunohistochemical staining of paraffin sections with anti-ErbB-2 antibody through the heart region of 10.5 dpc wild-type (Right) and mutant embryos (Left). (b) Enlargement of a. The ErbB-2 expression is globally reduced, including over the trabeculae (tr) and the compact zone (c) of the mutant ventricle (v). (c) Immunohistochemical labeling with an anti-5-HT2B-specific antibody performed on cryostat sections of 10.5-dpc embryos illustrates the wild-type expression of 5-HT2B receptor in the trabecular and the compact zone of the heart but not in the endocardial cushions (ec). (d) Semiquantitative reverse transcription–PCR shows a strong reduction of ErbB-2 mRNA in 9.5-dpc mutant embryos whereas ErbB-4 and the ribosomal elongation factor EF1a expression in the same RNA preparation remains unchanged. (e) A Western blot analysis with an anti-ErbB-2 antibody demonstrates a reduction of ErbB-2 expression in total protein extract from 5-HT2B mutant embryos (from 9.5 to 11.5 dpc) (Upper). The platelet-derived growth factor receptor immunoreactivity in the same mutant embryo extracts remains unmodified (Lower). a, Atrium; α, anti-. (Bars for a and c = 250 μm, and b = 25 μm.)
Identification of factors controlling myocardial differentiation and proliferation is of great importance for understanding the pathogenesis of congenital heart diseases. Remarkably, embryonic heart defects reported in the mice lacking neuregulin or its receptors, ErbB-2 (HER-2/neu) and ErbB-4 are similar to those noted in 5-HT2B embryos (19). Expression studies showed that neuregulin is expressed in the endothelial cells of the endocardium and both ErbB-2 and ErbB-4 are localized into the ventricular wall of the myocardium (20). In ErbB-2 or ErbB-4 mutant mice, strong defects in ventricular trabeculation that are nearly identical to that seen in most severely affected 5-HT2B mutant embryos, trigger death at 10.5 dpc. This similarity suggested possible interaction between these two signaling pathways. To this purpose, we analyzed the ErbB-2 and ErbB-4 expression in 5-HT2B mutants. Interestingly, at 9.5 dpc, mutant embryos exhibit a significant reduction in ErbB-2 mRNA, whereas ErbB-4 mRNA expression is not altered. Immunohistochemical analysis revealed that the pattern of ErbB-2 expression in the wild-type heart (Fig. 3 a and b Right) is overlapping that of 5-HT2B receptor expression (Fig. 3c) but is significantly reduced in 5-HT2B mutant embryo hearts (Fig. 3 a and b Left). Moreover, Western blot analysis revealed a significant reduction in ErbB-2 protein levels in the mutant embryo extracts at 9.5 dpc (Fig. 3 d and e). However, another growth factor receptor expressed in heart at this period, such as the platelet-derived growth factor receptor protein remains unchanged (Fig. 3e). Reduction in ErbB-2 protein expression becomes less significant at 11.5 dpc, i.e., after the period of lethality, because the most affected 5-HT2B mutant embryos are resorbed at 11 dpc (Fig. 3e). Because ErbB-2 can be phosphorylated (transactivated) by activation of G protein-coupled receptors (21), this cardiac phenotype could result from a lack of ErbB-2 transactivation by 5-HT2B. However, an ErbB-2 regulation at the transcriptional level seems more likely because both mRNA and protein levels of ErbB-2 are reduced in 5-HT2B mutant embryos. Moreover, in mouse LMTK− cells transfected by the 5-HT2B cDNA, 5-HT raises the endogenous expression but not the phosphorylation levels of ErbB-2 protein (data not shown). Furthermore, the 5-HT2B receptor expression remains unchanged in ErbB-2 knockout mice (22). We also investigated if ablation of 5-HT2B receptor selectively reduced expression of other genes that are involved in heart development. We performed reverse transcription–PCR as a quantitative approach for the expression of genes whose ablation resulted in defects in the heart, such as N-myc or for chamber-specific markers such as MLC2v. No significant changes in either marker (N-myc or MLC2v) mRNA expression were detected in the 5-HT2B mutant embryos (data not shown). The mechanism whereby 5-HT2B activation regulates ErbB-2 expression is currently under investigation in our lab. Because ErbB-2 but not ErbB-4 receptor levels are impaired in the 5-HT2B mutant mice, our data suggest that Gq-coupled 5-HT2B receptor specifically uses ErbB-2 receptor tyrosine kinase pathways in the heart.
Interestingly, mice lacking both Gαq and Gα11 also die at embryonic day 11 because of hypoplasia in the ventricular wall. Two active alleles of these genes are required for extrauterine life, and at least one intact Gαq allele is needed to bring the embryo to term (23). Gα11 seems to be preferentially required for 5-HT2B-dependent heart developmental events, because none of the skeletal abnormalities observed in Gαq−/−Gα11+/− embryos could be detected in the 5-HT2B mutants (data not shown).
The less affected 5-HT2B mutant embryos survived into the neonatal period and appeared normal in their gross morphology although a second period of lethality appears during the first week of life (Fig. 1e). Furthermore, 5-HT2B mutant mice fertility was consistently lower than the control 129/PAS strain, and their body weight was slightly reduced by 9%. Moreover, their percent of heart weight to body weight was significantly reduced by 28% (1.06 ± 0.12 vs. 0.76 ± 0.07; P < 0.05 by a Fisher test; n > 5). Investigation of newborn mutant hearts at histopathological level revealed a strong decrease in ventricular mass and abnormal compact layer in all observed newborn mutant hearts (n = 6) (Fig. 4a). Shortly after birth, cardiac myocytes lose their proliferative capacity, and growth of the myocardium occurs through enlargement of existing myocardial cells. A report indicated that 5-HT may regulate proliferation in the embryonic heart (8). We determined the in vivo proliferative capacity of myocardium in 5-HT2B newborn heart by measuring BrdUrd incorporation. Essentially, no BrdUrd incorporation was detected in the heart of 5-HT2B mutant newborn (Fig. 4c Left), whereas BrdUrd incorporation was clearly observed in the compact zone of the wild-type newborn heart (Fig. 4c Right). The hypoplasia in the 5-HT2B mutant heart is probably caused by impaired proliferation.
Figure 4.
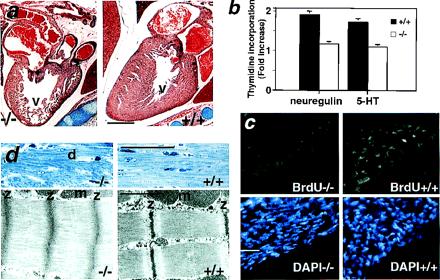
Heart defects in postnatal 5-HT2B mutant mice. (a) Histological analysis of newborn mutant hearts shows an obvious loss of ventricular (v) mass (hypoplasia). (b) Thymidine incorporation of isolated cardiomyocytes from newborn heart in response to 5-HT and neuregulin (heregulin) is significantly reduced in the 5-HT2B mutant, indicating that myocyte proliferation is impaired and this effect is cell autonomous. The thymidine incorporation rate is expressed as fold increase over basal ± SEM (−/−, n = 3; and +/+, n = 3). (c) In vivo, BrdUrd incorporation (green) is almost undetectable in the heart of 5-HT2B newborn mutant (Left), whereas BrdUrd incorporation was observed in the compact zone of control newborn heart (Right). Staining with 4′,6-diamidino-2-phenylindole of the same sections localizes the cell nuclei (blue). (d Upper) Semithin sections of heart in 6-week-old mutant shows abnormal morphology, including degenerating fiber (d) (Left). (d Lower) TEM of ultrathin sections shows that, in mutant heart, the sarcomere lengths are greatly reduced, the M-lines and the I-bands are indistinguishable, and the A-bands occupy the entire length between Z-bands (z) which themselves are thickened (Left) compared with wild-type age-mated heart (Right). m, Mitochondria. (Bars for a = 100 μm; c and d Upper = 20 μm; and d Lower = 0.5 μm.)
Accordingly, in isolated newborn ventricular myocytes from 5-HT2B mutant, thymidine incorporation in response to 5-HT was also reduced (Fig. 4b). These in vitro data indicate that the 5-HT2B mutant cardiac phenotype is intrinsic to myocytes and is cell autonomous. These data suggest that 5-HT indeed acts as a cardiac mitogenic factor via a 5-HT2B receptor-mediated pathway. Furthermore, the requirement for the neuregulin-ErbB-2 signaling pathways is demonstrated by a thymidine incorporation assay in response to neuregulin (heregulin) in the isolated cardiomyocytes from embryo (9.5 dpc) and newborn mice. Fig. 4b shows that 5-HT2B-deficient cardiomyocytes obtained from newborn hearts also have defective proliferative responses to neuregulin. These data strongly suggest that Gq-coupled 5-HT2B receptor specifically uses the receptor tyrosine kinase ErbB-2 signaling pathways in cardiac development.
Adult hearts from surviving 5-HT2B mutants (6 weeks old) also exhibited dilated chambers and a decreased ventricular mass, their percent of heart weight to body weight was significantly reduced by 24% (0.67 ± 0.4 vs. 0.51 ± 0.03; P < 0.05 by a Fisher test; n = 10). On histological sections, 6-week-old adult hearts from 5-HT2B mutants showed a consistent pattern of myocellular disorganization, and scattered areas of myocellular disarray unlike wild-type hearts (Fig. 4d Upper). At the TEM level, the mutant myocardium displays an irregular array of sarcomeric myofibrils including abnormally wide Z bands relative to controls (Fig. 4d Lower). In particular, the well-defined sarcomeric pattern is disrupted, the sarcomere lengths are greatly reduced, and M bands are indistinct. There were no obvious apoptotic bodies observed at the TEM level in the mutant heart either at 9.5 dpc, newborn, or adult stage. These cardiac sarcomeric defects are similar to those observed in mice heterozygous for the α–myosin heavy chain knockout (24) with numerous degenerating fibers. Loss of myofibrils is the most obvious structural change in many cardiomyopathies (25) and sarcomeric disarray is characteristic of failing hearts (26). Several studies have suggested a role for Gq-coupled receptors in various heart compartments (27) and Gq has been shown to regulate cardiomyopathy development. For example, both the angiotensin II receptor and α1B-adrenergic receptors signal via Gq. The angiotensin II transgenic mice died in utero or shortly after birth with grossly enlarged atria (28), but with no changes in ventricular morphology seen in α1B transgenic mice (29), indicating a specificity of downstream pathways. Parallel to the previous observations, targeted inhibition of Gq signaling by overexpression of a carboxyl-terminal peptide of Gq in the heart reduces ventricular hypertrophy in response to pressure overload (30), whereas D'Angelo et al. (31) found that a 4-fold overexpression of the Gq protein in the heart generates cardiac hypertrophy and heart failure.
We show that the 5-HT2B receptor is required for 5-HT to regulate cardiovascular functions. Previously, we have shown that treatment of embryos with selective antagonists of 5-HT2B receptor caused trabeculation defects in the heart along with defects in neural crest cell derivatives. 5-HT2B mutant mice exhibit obvious cardiac defect but the gross morphology of the CNS appears unaffected. These preliminary observations suggest that the morphological effects of antagonists on neural crest cell derivatives in embryo cultures are probably masked in the 5-HT2B mutant mice, but could be reflected at functional levels. Further investigations are necessary to assess if CNS functions are affected by 5-HT2B receptor elimination. The present data strongly suggest that the Gq-coupled-5-HT2B receptor is involved in cardiac proliferation and differentiation. These findings should facilitate a genetic approach and a new drug design in the cardiovascular disease.
Acknowledgments
We thank Drs. K. Niederreither, F. Guillemot, and Prof. M. Mark for stimulating discussions and critical reading of the manuscript. This work was supported by funds from the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, the Hôpital Universitaire de Strasbourg, the Université Louis Pasteur, and by grants from the Association Française contre les Myopathies, Association pour la Recherche contre le Cancer, the Ligue Nationale contre le Cancer, and a Lilly-University L. Pasteur Fellowship (C.G.N.).
Abbreviations
- CNS
central nervous system
- dpc
day postcoitum
- ES
embryonic stem
- 5-HT
serotonin (5-hydroxytryptamine)
- TEM
transmission electron microscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wilkinson L O, Dourish C T. In: Serotonin Receptor Subtypes: Basic and Clinical Aspects. Peroutka S J, editor. Vol. 15. New York: Wiley–Liss; 1991. pp. 147–210. [Google Scholar]
- 2.Saxena P R, Villalon C. Trends Pharmacol Sci. 1991;12:223–227. doi: 10.1016/0165-6147(91)90556-8. [DOI] [PubMed] [Google Scholar]
- 3.Wade P R, Chen J, Jaffe B, Kassem I S, Blakely R D, Gershon M D. J Neurosci. 1996;16:2352–2364. doi: 10.1523/JNEUROSCI.16-07-02352.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham-Smith D. The Carcinoid Syndrome. London: W. Heinemann; 1972. [Google Scholar]
- 5.Lauder J M, Wallace J A, Krebs H. Adv Exp Med Biol. 1981;133:477–506. doi: 10.1007/978-1-4684-3860-4_28. [DOI] [PubMed] [Google Scholar]
- 6.Lauder J M. Prog Brain Res. 1988;73:365–387. doi: 10.1016/S0079-6123(08)60516-6. [DOI] [PubMed] [Google Scholar]
- 7.Colas J-F, Launay J-M, Vonesch J-L, Hickel P, Maroteaux L. Mech Dev. 1999;87:77–91. doi: 10.1016/s0925-4773(99)00141-0. [DOI] [PubMed] [Google Scholar]
- 8.Yavarone M S, Shuey D L, Tamir H, Sadler T W, Lauder J M. Teratology. 1993;47:573–584. doi: 10.1002/tera.1420470609. [DOI] [PubMed] [Google Scholar]
- 9.Hoyer D, Fozard J R, Saxena P R, Mylecharane E J, Clarke D E, Martin G R, Humphrey P P A. Pharmacol Rev. 1994;46:157–203. [PubMed] [Google Scholar]
- 10.Murphy D L, Wichems C, Li Q, Heils A. Trends Pharmacol Sci. 1999;20:246–252. doi: 10.1016/s0165-6147(99)01325-5. [DOI] [PubMed] [Google Scholar]
- 11.Choi D-S, Ward S, Messaddeq N, Launay J-M, Maroteaux L. Development (Cambridge, UK) 1997;124:1745–1755. doi: 10.1242/dev.124.9.1745. [DOI] [PubMed] [Google Scholar]
- 12.Choi D-S, Maroteaux L. FEBS Lett. 1996;391:45–51. doi: 10.1016/0014-5793(96)00695-3. [DOI] [PubMed] [Google Scholar]
- 13.Tournois C, Mutel V, Manivet P, Launay J M, Kellermann O. J Biol Chem. 1998;273:17498–17503. doi: 10.1074/jbc.273.28.17498. [DOI] [PubMed] [Google Scholar]
- 14.Manivet P, Mouillet-Richard S, Callebert J, Nebigil C G, Maroteaux L, Hosoda S, Kellermann O, Launay J-M. J Biol Chem. 2000;275:9324–9331. doi: 10.1074/jbc.275.13.9324. [DOI] [PubMed] [Google Scholar]
- 15.Launay J-M, Birraux G, Bondoux D, Callebert J, Choi D-S, Loric S, Maroteaux L. J Biol Chem. 1996;271:3141–3147. doi: 10.1074/jbc.271.6.3141. [DOI] [PubMed] [Google Scholar]
- 16.Nebigil C G, Launay J-M, Hickel P, Tournois C, Maroteaux L. Proc Natl Acad Sci USA. 2000;97:2591–2596. doi: 10.1073/pnas.050282397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams J W, Sakata Y, Davis M G, Sah V P, Wang Y, Liggett S B, Chien K R, Brown J H, Dorn G W, II. Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux C, Madani M, Launay J-M, Rey F, Citadelle D, Mulliez N, Kolf M. Toxicol in Vitro. 1995;9:653–662. doi: 10.1016/0887-2333(95)00071-f. [DOI] [PubMed] [Google Scholar]
- 19.Lemke G. Mol Cell Neurosci. 1996;7:247–262. doi: 10.1006/mcne.1996.0019. [DOI] [PubMed] [Google Scholar]
- 20.Lee K F, Simon H, Chen H, Bates B, Hung M C, Hauser C. Nature (London) 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 21.Daub H, Weiss F U, Wallasch C, Ullrich A. Nature (London) 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 22.Morris J K, Lin W, Hauser C, Marchuk Y, Getman D, Lee K F. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- 23.Offermanns S, Zhao L P, Gohla A, Sarosi I, Simon M I, Wilkie T M. EMBO J. 1998;17:4304–4312. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones W K, Grupp I L, Doetschman T, Grupp G, Osinska H, Hewett T E, Boivin G, Gulick J, Ng W A, Robbins J. J Clin Invest. 1996;98:1906–1917. doi: 10.1172/JCI118992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mann D L, Urabe Y, Kent R L, Vinciguerra S, Cooper G T. Circ Res. 1991;68:402–415. doi: 10.1161/01.res.68.2.402. [DOI] [PubMed] [Google Scholar]
- 26.Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, Friedl A, Bleese N. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- 27.McKinsey T A, Olson E N. Curr Opin Genet Dev. 1999;9:267–274. doi: 10.1016/s0959-437x(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 28.Hein L, Barsh G S, Pratt R E, Dzau V J, Kobilka B K. Nature (London) 1995;377:744–747. doi: 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- 29.Milano C A, Dolber P C, Rockman H A, Bond R A, Venable M E, Allen L F, Lefkowitz R J. Proc Natl Acad Sci USA. 1994;91:10109–10113. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akhter S A, Luttrell L M, Rockman H A, Iaccarino G, Lefkowitz R J, Koch W J. Science. 1998;280:574–577. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 31.D'Angelo D D, Sakata Y, Lorenz J N, Boivin G P, Walsh R A, Liggett S B, Dorn G W, II. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]