

Abstract
At the onset of Drosophila metamorphosis, the steroid hormone 20-OH ecdysone directly induces a small number of early puffs in the polytene chromosomes of the larval salivary gland. Proteins encoded by the early genes corresponding to these transcriptional puffs then regulate the activity of both the early puffs themselves and a much larger set of late puffs. Three of these early genes encode transcription factors that play critical regulatory roles during metamorphosis. Here we report the cloning, DNA sequence, genomic structure, ecdysone inducibility, and temporal expression of an early gene residing in the 23E early puff and denoted E23 (Early gene at 23). In contrast to other early genes, E23 encodes a protein with similarity to ATP-binding cassette transporters. Using heat shock-inducible transgenes, we found that E23 overexpression not only produces phenotypic abnormalities and lethality, but also interferes with ecdysone-mediated gene activation, demonstrating that E23 is capable of modulating the ecdysone response. Our results suggest the existence of a previously unrecognized regulatory mechanism for modulating steroid hormone signaling in Drosophila.
The fruitfly Drosophila melanogaster provides a powerful model system for elucidating the molecular and genetic mechanisms by which steroid hormones regulate developmental processes. In Drosophila, changes in the concentration of the steroid hormone 20-OH ecdysone (hereafter referred to as ecdysone) regulate specific developmental events, the most dramatic of which is the metamorphosis from the larval to the adult form (1). The early stages of metamorphosis are coordinated by three discrete ecdysone pulses. The late-larval ecdysone pulse triggers metamorphosis and leads to puparium formation (pupariation) some 6 h later, transforming the feeding larva into an immobile white prepupa. Immediately after pupariation, the whole-body ecdysone concentration rapidly declines to a low level by the mid-prepupal stage, followed by a second, prepupal ecdysone pulse that peaks 10 h after pupariation. Head eversion occurs 2 h later, giving rise to a fully formed pupa. Pupation then is followed by a broad pupal ecdysone pulse that is necessary for tissue differentiation and adult development.
Drastic changes in the body plan of the organism occur during metamorphosis. Most of the strictly larval tissues are eliminated through histolysis, involving both programmed cell death and phagocytosis. Other larval tissues, such as the central nervous system, are extensively remodeled (2), whereas the imaginal tissues undergo morphogenesis and differentiate to produce most of the adult structures (3). Although it is clear that the fluctuations in ecdysone concentration are required for the coordination and proper execution of these tissue-specific developmental programs, little is known about how hormone concentrations are regulated or how different tissue and cell types respond to the hormonal signal in different ways.
This hormonal signal is transduced primarily through the action of an ecdysone receptor that binds both DNA and hormone as a heterodimer of two nuclear receptor proteins encoded by the ecdysone receptor (EcR) and ultraspiracle (usp, the Drosophila RXR gene) genes (4, 5). The EcR gene encodes three protein isoforms (EcR-A, EcR-B1, and EcR-B2), that are differentially expressed in tissues with different hormone responses (6). Although all three isoforms contain the same DNA-binding domain, functional differences between the isoforms may contribute to the cellular and tissue specificity of the hormone response (7). Formation of a functional ecdysone receptor also requires the activity of a complex of molecular chaperones (8).
Underlying the different responses to ecdysone during metamorphosis are changes in gene expression, most recently demonstrated on a genomewide scale with DNA microarrays (9). Coordinate transcriptional activation and repression of specific genes during the early stages of metamorphosis were first observed as the induction and regression of transcriptional puffs in the polytene chromosomes of the late third-instar larval salivary gland. Experimental analysis of puffing in cultured salivary glands led to a hierarchical model for the genetic regulation of the late larval ecdysone response (10). In this model, a set of 5–6 early puffs directly induced by ecdysone were predicted to encode regulatory molecules necessary for the activation of a much larger set of late genes (>100) and for the repression of the early genes themselves. Cloning of the genes corresponding to three of these early puffs confirmed and extended this hierarchical model by showing that their genes contained transcription units that exhibited a primary response to ecdysone and encoded transcription factors. Thus, the E74 gene encodes two members of the ETS oncogene family (isoforms E74A and B); the E75 gene encodes three members of the steroid-receptor superfamily (isoforms E75A, B, and C); and the Broad-Complex (BR-C) encodes a family of transcription factors containing TFIIIA-like zinc fingers (see ref. 1 and references therein). Further molecular-genetic analysis has revealed that these early genes provide critical regulatory functions during metamorphosis, and both E74- and BR-C-encoded transcription factors have been shown to regulate gene expression through direct binding to DNA sequences in specific target genes (11).
Given the importance of the early genes identified within the previously characterized early puff loci, we set out to clone the gene(s) corresponding to the early puff at cytological location 23E, and we report here our initial identification and characterization of the E23 early gene. Surprisingly, we have found that the E23 gene encodes a protein with homology to ATP-binding cassette (ABC) transporters, a diverse family of transmembrane transport proteins that function independently of the Golgi apparatus and the classical secretory pathway (12, 13). Mutations in ABC transporter genes are associated with a number of inherited human disorders, including cystic fibrosis, adrenoleukodystrophy, Stangardt's disease, progressive familial intrahepatic cholestasis, and Tangier's disease (14, 15).
The analysis of wild-type and ectopic E23 expression reported here shows that the E23 early gene is capable of regulating ecdysone responses during metamorphosis. We discuss possible mechanisms by which this regulation might occur and consider the implications of these results for the regulation of hormone responses during Drosophila metamorphosis.
Materials and Methods
Construction and Analysis of the 23E Contig.
Purified DNA from yeast artificial chromosome DY469 (16) was used to identify genomic clones containing primarily single-copy sequences from the 23DE region and those producing overlapping restriction-digestion patterns cross-hybridized to produce a contig. This contig was extended proximally across the 23E puff region by chromosome walking, and the genomic clones mapped relative to the 23E puff by in situ hybridization. Additional clones (cosA2, cosEC5, P1-D) were generously provided by R. Kelley (Baylor College of Medicine, Houston), J. Lucchesi (Emory University, Atlanta), and B. Baker (Stanford University, Stanford, CA).
Differential cDNA Hybridization.
Preparation of radio-labeled cDNA probes was carried out essentially as described by Stowers et al. (17). Four identical Southern blots containing EcoRI and SalI restriction-digested genomic clone DNA from the 23E contig were prepared and hybridized with cDNA probes synthesized from poly(A)+ mRNA of late third-instar larvae before vs. after the metamorphic ecdysone pulse and from poly(A)+ mRNA of late third-instar larval tissues cultured in insect-tissue culture media with vs. without 20-OH ecdysone (5 × 10−6 M) for 5–6 h.
Identification of E23 cDNAs and Determination of E23 Gene Structure.
We identified E23 cDNAs by screening a λgt10 phage library constructed from late third-instar larval tissues cultured in the presence of cycloheximide plus ecdysone (a gift of C. Thummel, University of Utah, Salt Lake City) and a directional cDNA library constructed from 0- to 2-h prepupae (17) with the genomic restriction fragment identified by differential hybridization.
E23-c1-derived deletion subclones were generated by means of an Exo-Size Deletion kit (New England Biolabs) and sequenced. The partial sequences obtained were used to determine the sequence of gene-specific primers that were used to complete the cDNA sequence on both strands.
An E23 transcription start site was determined to be 18 bp upstream of the 5′ end of the clone by rapid amplification of cDNA ends (RACE) and primer extension analysis. 5′ RACE was performed with a primer from the 5′ untranslated region (5′-GAGCTATTCCGCTTTTCCAC-3′) using the Marathon cDNA amplification kit (CLONTECH). PCR products were purified (Qiagen, Chatsworth, CA) and cloned with the TA-cloning kit (Invitrogen), and the eight longest clones were sequenced. Primer extension was performed with the same primer by using an X-tend kit (GIBCO/BRL). The resulting primer extension product was compared to the sequencing ladder produced with the same primer to sequence a 3.2-kb genomic PstI fragment containing the putative transcriptional start site. Because the E23-c1 cDNA lacked a poly(A)+ tail, we sequenced three additional E23-derived cDNAs and identified a poly(A)+ addition site 10 bp downstream of the 3′ end of the E23-c1 clone.
Genomic restriction fragments in the 23E contig containing E23 exons were identified by hybridization with E23-c1 probes, subcloned into pBluescript KS (Stratagene), and sequenced with vector and gene-specific primers.
Developmental Staging of Animals.
Staged embryos were obtained from large-population cages of wild-type Oregon R flies. All larvae raised for staging were maintained at 25°C on instant Drosophila medium (Carolina Biological Supply) supplemented with live yeast. First- and second-instar larvae were obtained by aging of 0- to 12-h embryo collections. L1 larvae were harvested by flotation in 30% glycerol and 1.5 M NaCl; salt was omitted for collections of L2 and L3 larvae. Larvae were rinsed and either flash-frozen in liquid nitrogen for storage at −70°C or used immediately for the isolation of total RNA. Third-instar larvae were staged by aging of 0- to 4-h-old L3 larvae collected at the L2/L3 molt. Prepupal and pupal time points were obtained by collection of white prepupae every 30 min for 4 h (eight collections in all), and allowed to develop for the number of hours required. Three animals from each timed collection (24 in all) were combined and used for RNA isolation. Prepupae staged at 2-h intervals were obtained by collection of white prepupae and then aging them for the number of hours shown before RNA isolation.
For isolation of larval tissues before dissection, staging of late third-instar larvae was performed as described by Andres and Thummel (18). In brief, larvae were cultured on standard or instant fly medium (Carolina Biological Supply) containing 0.05% bromophenol blue and were staged on the basis of disappearance of the blue dye from their guts. Larvae whose guts were completely blue were approximately 18 h from pupariation; those whose guts had cleared the blue dye after the cessation of feeding were approximately 4 h from pupariation.
RNA Isolation and Northern Hybridization.
Total RNA was isolated as described by Andres and Thummel (18), and Northern blot hybridizations was carried out essentially as described by Karim and Thummel (19). A 900-bp EcoRV fragment from the E23-c1 cDNA was cloned into pBluescriptKS (Stratagene), and 300–500 ng of the resulting recombinant plasmid was linearized with HindIII and used for the synthesis of E23-derived riboprobes.
Isolation and in Vitro Culture of Larval Tissues.
Late third-instar larvae before the late larval ecdysone pulse were identified as above and hand dissected in Grace's insect-tissue culture medium (GIBCO/BRL) supplemented with 2% ethanol at 5:1 (20). Larval tissues were incubated in 6-well tissue-culture plates with 1 ml of medium per well for 1 h in the absence of hormone. This medium then was removed and replaced with fresh medium with or without ecdysone at 5 × 10-6 M (Sigma) only and with or without ecdysone in the presence of cycloheximide (70 μg/ml final). After incubation for the required period, tissues were collected by centrifugation and total RNA was extracted for Northern blot analysis.
We distinguished transgenic and nontransgenic larvae by using yellow as a larval marker. For the B1 transgene on the X chromosome, y+ w P{hs:E23}B1 males were crossed with yw;P{6XEcRE:LacZ}2 females, generating y+ female larvae bearing one copy of the hs:E23 transgene and one copy of the LacZ transgene and y− male larvae bearing only the LacZ transgene. Similarly, for the J3 transgene on the third chromosome, we crossed yw;P{hs:E23}J3/TM6, y+ females with yw;P{6XEcRE:LacZ}2 males, in this case generating y− larvae that bore the hs:E23 transgene and y+ larvae that did not. Larvae were collected (≈18 h before pupariation) and dissected in Grace's insect-tissue culture medium, given either a 20-min heat shock (P{hs:E23}B1) or a 45-min heat shock (P{hs:E23}J3) at 37°C, and allowed to recover at 25°C for 1 h. These tissues then were incubated in fresh medium containing ecdysone (5 × 10−6 M) for 2, 4, or 6 h before isolation of total RNA for Northern blots.
Construction and Transformation of hs:E23 Transgene.
The hs:E23 transformation vector was constructed by purification of a 3.1-kb SacI–PstI fragment from the E23-c1 cDNA containing the entire ORF of E23, repair of the 3′ overhangs with T4 DNA polymerase, and ligation of the resulting fragment into the unique HpaI site of pCasperHS. Transformation was performed essentially as described by Rubin and Spradling (21).
Results
Identification of the E23 Early Gene Within the 23E Early Puff.
To identify early ecdysone-response genes within the 23E puff, we first constructed a genomic contig spanning the puff, shown in Fig. 1. In situ hybridization signals obtained by using cosmids 4.3 and EC5 bracket the 23E puff, demonstrating that the contig spanned the entire 23E puff (Fig. 1 Inset) and therefore was likely to contain the transcription unit(s) whose expression is correlated with puffing. We next identified a genomic restriction fragment containing exons whose transcription was induced by the late third-instar ecdysone pulse and whose transcription depended on the presence of ecdysone in larval tissues cultured in vitro. This fragment was used to identify >20 independent cDNAs, all of which were derived from a single transcription unit spanning >20 kb of genomic DNA. On the basis of transcriptional analyses presented below, the gene corresponding to this transcription unit has been designated E23 (Early gene at 23).
Figure 1.
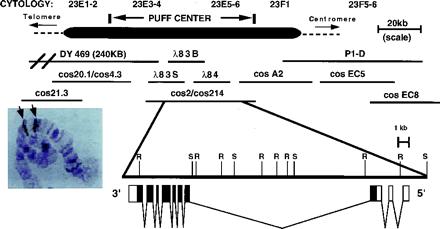
Genomic organization of the E23 locus, showing the 23E puff region (Top) and the corresponding genomic region (Middle). In situ hybridization results obtained with cosmid 21.3 and cosmid EC5 probes (Inset photo) demonstrate that the genomic contig spans the 23E puff. The genomic structure of the E23 transcription unit also is shown (Bottom) and includes a restriction map of the genomic region (r = EcoRI, S = SalI) and the position and size of the E23 exons. Noncoding regions are shown as open boxes and coding regions as solid boxes. Two largely overlapping cosmid pairs, cos20.1/cos4.3 and cos2/cos214, are shown as a single contiguous line.
The longest E23-derived cDNA identified (E23-c1) was sequenced in its entirety and the 5′ and 3′ ends of an E23 transcription unit determined. Comparison of the resulting composite E23 cDNA sequence (AF270979) with the DNA sequence determined for the corresponding genomic regions showed that the identified E23 transcription unit contains 10 exons spanning >20 kb of genomic DNA (Fig. 1).
E23 Encodes a Member of the ABC Transporter Family.
The E23-c1 cDNA contains an ATG-initiated ORF beginning in the third exon and terminating in the tenth and final exon that gives a predicted 95-kDa polypeptide of 856 aa. Comparison to protein databases shows that E23 is a member of the superfamily of ABC transporters (Fig. 2), containing a highly conserved nucleotide-binding domain (NBD) as well as a less-conserved region of hydrophobic domains corresponding to putative transmembrane domains (TMs) found in such proteins (22). An amino acid alignment is shown in Fig. 2 for a conserved region of the putative E23 protein (234 aa in length) that includes the ABC. Functional transporters are composed of two halves, each containing an NBD and a TM domain, whereas the E23 gene encodes a putative half-transporter, containing one NBD and one set of TMs, and is likely to function as either a homodimer or as a heterodimer with other half-transporters. E23 is most similar to half-transporters from a variety of other organisms; in the ABC region shown, amino acid identity is 34–42% and amino acid similarity is 57–59% with the yeast ADP1, Drosophila ATET, Drosophila white, human ABCG1/murine ABC8/human White, and human ABCG2/MXR/ABCP1/BCRP (Fig. 2).
Figure 2.

Alignment of the ABC region of the putative E23 protein with other members of the ABC transporter family. Amino acids identical to those found in the putative E23 protein are shown as black boxes with white text, and those that are similar as gray boxes with black text. Thick lines over the E23 sequence show the location of the Walker A and B motifs and the location of the ABC signature. GenBank accession nos. are as follows: E23 (AF270979); ADP1 (NP009937); ABCG2 (AAC97367); White (GI1070660); ABCG1 (NP0033723); and ATET (AAF51027).
Expression of the E23 Early Gene.
The E23-c1 cDNAs hybridize with an RNA transcript of approximately 4.5 kb that is rapidly induced in larval tissues cultured in the presence of ecdysone (Fig. 3A). This ecdysone-mediated transcriptional activation occurs even in the presence of the protein synthesis inhibitor cycloheximide, defining the ecdysone-mediated transcriptional activation as a primary response (Fig. 3B).
Figure 3.
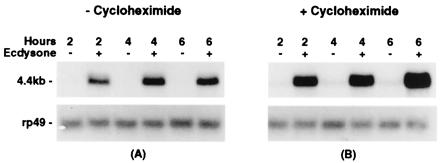
Ecdysone-regulation of E23 transcription. Shown are the results of hybridizing an E23-specific probe to Northern blots prepared with total RNA isolated from larval tissues cultured under the conditions shown. (A) E23 transcripts were detected in larval tissues cultured in the presence (+) but not in the absence (−) of ecdysone for 2, 4, and 6 h. Transcript accumulation reaches a maximum at ≈4 h and remains constant through 6 h. (B) E23 transcripts were detected in larval tissues cultured in the presence of both ecdysone and cycloheximide (+) but were not detected in larval tissues cultured in the presence of cycloheximide only (−). The RP49 gene encodes a ribosomal protein and is used as a control for RNA loading.
Determination of the temporal pattern of E23 transcript accumulation during development demonstrates that E23 expression corresponds with most of the observed developmental pulses of ecdysone (Fig. 4), consistent with ecdysone induction of E23 expression during development.
Figure 4.
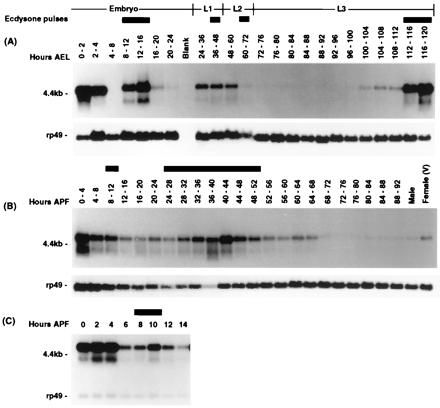
Developmental profile of E23 transcription. Developmental Northern blots were prepared with total RNA isolated from animals at the developmental times indicated and hybridized with an E23-specific probe (see Materials and Methods). The hours and stages of development, as well as the developmental periods associated with ecdysone pulses, are indicated above the lanes. (A) Developmental time points are indicated with respect to hours after egg laying and stages include embryonic development (embryo), first-instar (L1), second-instar (L2), and third-instar (L3) larval stages. (B) Developmental time points are indicated with respect to hours after pupariation. (C) Developmental time points taken at 2-h intervals after pupariation through 14 h. Hybridization results obtained with an RP49 probe as a loading control are shown below each lane in A and B. The prepupal Northern shown in C was simultaneously hybridized with both probes simultaneously.
Several features of E23 developmental expression appear unique to E23 in comparison to other characterized early genes (23–26). First, elevated levels of E23 mRNA found in females and in 0- to 2-h embryos suggests E23 transcripts are maternally deposited; this is not seen for other early genes. Second, E74 and E75 are expressed late in embryogenesis (20–24 h after egg laying) at a developmental time that does not correspond to a recognized ecdysone pulse; this late embryonic expression is not observed for E23. Last, although the abundance of early gene transcripts typically decreases rapidly after pupariation, reappearing in a narrow burst induced by the prepupal ecdysone pulse, E23 transcripts remain abundant for a longer period after pupariation before decreasing and rising in apparent response to the prepupal ecdysone pulse at 10 h after pupariation.
Phenotypes Associated with Ectopic Expression of E23.
To investigate the function of the E23 gene, we examined the phenotypic consequences of ectopic E23 expression during development by using animals bearing either an X-linked or an autosomal transgene (P{hs:E23}B1 and P{hs:E23}J3, respectively). We observed that the relative viability of transgenic flies was reduced to between 3% and 30% that seen for nontransgenic sibs after a 1-h, 37°C heat shock each day during development. Additionally, most of the surviving adults had reduced, thin thoracic bristles, and 5–10% also had malformations of the third pair of legs (data not shown). Neither mutant phenotype was observed in nontransgenic siblings, indicating that these phenotypes result from ectopic expression of E23. In contrast, a 1-h, 37°C heat shock every 12 h through development was lethal to all transgenic animals, with lethality occurring primarily during the early stages of metamorphosis.
The observation that some transgenic animals survive a 1-h heat shock once a day but not twice a day, and the identification of metamorphosis as the predominant lethal phase associated with ectopic E23 expression, suggested that the developmental timing and/or amount of ectopic E23 expression are important determinants for the observed phenotypic responses. To examine this further, we performed single, 37°C, 1-h heat shocks at specific developmental stages before and during metamorphosis (Table 1). Heat-shock induction of E23 expression just before the late-larval ecdysone pulse that triggers metamorphosis resulted in the death of 60% of the P{hs:E23}B1 animals. However, a single heat shock administered to larvae during the late-larval ecdysone pulse, either 4 h before pupariation or at pupariation, resulted in complete lethality (100%), with little further developmental progression. Heat shock during the mid-prepupal stage or during the prepupal ecdysone pulse causes the death of most, although not all of the heat-shocked animals, whereas overexpression induced by later heat shock was associated with only moderate reductions in viability (Table 1). In contrast, most or all P{hs:E23}B1 transgenic animals survived in the absence of heat shock, as did heat-shocked control w1118 larvae, prepupae, and pupae.
Table 1.
Reductions in viability associated with a single heat shock-induced pulse of E23 expression during late larval and prepupal development
| Developmental stage at heat shock | Total no. of animals | No. of enclosed adults | Viability, % |
|---|---|---|---|
| Late L3 larvae (≈18 hrs before pupariation) | 52 | 21 | 40.3 |
| Late L3 larvae (≈4 hrs before pupariation) | 84 | 0 | 0.0 |
| White prepupae (0 hrs before pupariation) | 73 | 0 | 0.0 |
| 4–6 h after pupariation | 73 | 16 | 21.9 |
| 8–10 h after pupariation | 59 | 13 | 22.0 |
| 12–14 h after pupariation | 70 | 50 | 71.4 |
| 24 h after pupariation | 75 | 57 | 76.0 |
| 30 h after pupariation | 56 | 33 | 58.9 |
Ectopic Expression of E23 Suppresses Hormone-Mediated Transcriptional Activation.
These phenotypic results suggested that ectopic E23 expression interferes with steroid-hormone signal transduction. To test this hypothesis, we examined the effect of ectopic E23 expression on hormone-mediated transcriptional activation by using in vitro-cultured larval tissues (Fig. 5). In larval tissues derived from nontransgenic animals (−), we observe hormone-mediated transcriptional activation, although we note a slight delay in this response associated with heat shock (compare Figs. 3A and 5A). In larval tissues from transgenic sibling animals (+), significant transcriptional activation was not observed even after 6 h of culture in the presence of hormone. This loss of transcriptional activation was seen with both the X-linked transgene (Fig. 5A) and the autosomal transgene (Fig. 5B) for the hs:E23 transgene, and affected a number of ecdysone-inducible genes, including E74, E75, and E23 early genes, as well as an ecdysone-inducible LacZ transgene (P{6XEcRE:LacZ}2,4) containing a minimal promoter driven by six copies of the ecdysone receptor response element found in the hsp27 gene (4), but had no apparent effect on rp49 transcript accumulation. These results demonstrate that ectopic expression of E23 interferes with steroid hormone-mediated transcriptional activation.
Figure 5.
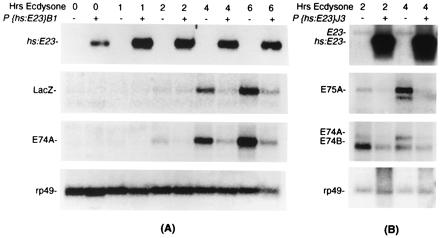
Suppression of ecdysone-mediated transcription by ectopic E23 expression. Tissues isolated either from larvae bearing a hs:E23 transgene (+) or from sibling larvae lacking this transgene (−) were subjected to heat shock (30 min) and cultured in the presence of 5 × 10−6 M ecdysone for the number of hours indicated, after which total RNA was isolated and used to produce Northern blots hybridized with gene-specific probes as shown. Representative results are shown for both the P{hs:E23}B1 transgene (A) and the P{hs:E23}J3 transgene (B).
Discussion
The E23 Early Gene Encodes an Ecdysone-Induced ABC Transporter.
We have identified the early ecdysone-response gene E23 within the 23E early puff. E23 expression occurs as a primary response to ecdysone and is correlated with pulses of ecdysone during development, all characteristics it shares with other early genes in the ecdysone genetic regulatory hierarchy. We note, however, that E23 may not be the only ecdysone-responsive ABC transporter gene in the 23E puff region. Preliminary analysis of nearby ABC transporter-coding regions identified through the recently released Drosophila genome sequence (27) suggests that transcription of one of these coding regions is also ecdysone regulated (B. Ring and D.G., unpublished results).
Expression of a number of ABC transporter genes in other organisms also has been shown to be up-regulated by steroid hormones, including the multidrug resistance genes in mice and in mammalian cell lines (28–30). Most recently, the human white/ABC8/ABCG1 half-transporter gene (Fig. 2) was shown to be directly induced by oxysterols such as 25-hydroxycholesterol; this transcriptional activation also required the activity of specific nuclear receptors (31). The ABCG1 gene and a number of other human ABC transporter genes display up-regulation in response to cholesterol (acetylated low density lipoprotein) loading in human macrophages (15, 32). These results suggest an involvement of ABC transporter genes in regulating and/or executing hormone responses.
The E23 Early Gene Is Capable of Acting as a Negative Regulator of Hormone Responses.
Three of the previously cloned early genes corresponding to early puff loci encode transcription factors that regulate downstream target genes by DNA binding, consistent with the expectations derived from the hierarchical model (11). Although it was therefore initially surprising to find that the E23 early gene encodes a member of the superfamily of ABC transporters, the consequences of ectopic E23 expression suggest that E23 acts as a negative regulator of ecdysone-mediated signaling. First, E23-induced lethality is developmentally restricted to the early stages of metamorphosis, suggesting that expression of the endogenous E23 gene must be carefully regulated during this period. Second, less severe heat-shock induction regimens produce flies that display mutant phenotypes similar to those associated with other mutations thought to cause reductions in hormone signal transduction (7, 8, 33). Third, ectopic expression of E23 is sufficient to block hormone-mediated gene activation of a number of early ecdysone response genes as well as a LacZ transgene whose expression is driven by ecdysone receptor response elements.
On the basis of these results, one likely in vivo function for E23 is to participate in the negative autoregulation of early genes during the late larval ecdysone pulse (14). This early-gene repression is accomplished in part through the action of a nuclear receptor encoded by another primary response gene, DHR3 (34, 35). However, ectopic DHR3 expression results in only a partial repression of early-gene expression, and conversely, early-gene repression is still observed in mutant prepupae expressing little or no DHR3, demonstrating that additional mechanisms for early gene repression must exist (36).
Steroids Are Substrates for ABC Transporters.
One speculative hypothesis consistent with our experimental results is that ectopic E23 reduces the effective concentration of ecdysone within the cell. This hypothesis is supported by data demonstrating that ABC transporters are capable of transporting steroids in a variety of organisms. In the yeast Saccharomyces cerevisiae, both the PDR5 and the SNQ2 transporters are capable of transporting steroids such as dexamethasone (37, 38), and mammalian P-glycoproteins have been shown to be capable of transporting cortisol, aldosterone, and dexamethasone (39–41). Most recently, mutations in the human ABC1 transporter gene have been shown to cause Tangier's disease and familial high-density lipoprotein deficiency, demonstrating a role for ABC1 in the regulation of intracellular cholesterol levels and cholesterol efflux (14). Interestingly, the murine ABC1 transporter is required in macrophages for the engulfment of dead cells, as is the Caenorhabditis elegans homolog, CED-7 (42). An involvement in cell death and apoptosis also has been postulated for the P-gp transporter because P-gp can protect mammalian cells from stimuli that induce caspase-mediated apoptosis (43). Caspases also have been implicated in a steroid-triggered cell-death response during metamorphosis in Drosophila (44), raising the possibility that one or more ABC transporters is involved in the cell death response in Drosophila.
Potential Regulation of Ecdysone Responses by E23-Mediated Modulation of Hormone Concentrations.
Modulation of hormone concentrations by an early gene encoding an ABC transporter would provide a mechanism for regulating hormone responses during metamorphosis and suggests a number of possible functions for the E23 gene. First, E23 expression would be expected to modulate the timing of early gene activation and repression during the late larval ecdysone response (45). If E23 transcriptional activation has an ecdysone dose-response similar to those of other early puff genes, then E23 would fine-tune the ecdysone response with respect to the ecdysone concentrations in the hemolymph, providing an elegant example of negative autoregulation. In conjunction with direct transcriptional regulators such as DHR3, the ecdysone-activated genes then could be turned off, and the relevant puffs would regress, while other, ecdysone-repressed genes would become active, and late puffs would be induced. Second, E23 could regulate tissue-specific differences in effective hormone concentrations, providing a mechanistic explanation for differences in the timing of early gene activation in different tissues and coordinating tissue-specific ecdysone responses (25, 46). Consistent with this idea, we find that E23 is dynamically expressed in a variety of tissues during metamorphosis (T.C. and D.G., unpublished results). Third, E23 may regulate hormone responses after the late larval ecdysone pulse, when a rapid decrease in whole body ecdysteroid concentrations occurs. This relatively hormone-free mid-prepupal period is required for the activation of specific puffs and their corresponding genes, including the βFTZF1 gene, encoding a nuclear receptor competence factor required for the prepupal ecdysone response (47, 48). Ecdysone dose-response experiments with cultured salivary glands have suggested that ecdysone concentrations as low as 5 × 10−9 M are sufficient to inhibit these mid-prepupal responses, but such low ecdysone concentrations have never been seen by radioimmune assay (47). E23 could provide a solution to this problem by lowering the effective ecdysone concentration within the relevant target tissues. Last, we note that E23 may participate more directly in regulating the whole body ecdysteroid levels by directing ecdysteroids into and/or out of specific tissues involved in such processes as metabolic inactivation and excretion of ecdysteroids, and the formation of ecdysteroid conjugates (see refs. 49 and 50).
Using the powerful genetic approaches available in Drosophila, we hope to determine the in vivo functions of E23 during Drosophila development and the mechanisms by which E23 modulates hormone responses. Comparison of our results to those obtained in other organisms suggests that ABC transporters are likely to play a role in hormone responses in most higher eucaryotes and that insights gained through the study of E23 and other ABC transporters in Drosophila are likely to be more generally applicable.
Acknowledgments
Special thanks to D. S. Hogness, S. Stowers, and K. White for assistance and support during the cloning of the E23 gene. We also thank L. von Kalm, C. Bayer, and C. Trivigno for fly transformations; B. Baker, G. Bashaw, R. Kelley, M. Kuroda, A. Pannutti, and J. Lucchessi for clones and unpublished data; P. Hurban and C. Thummel for E74 probes; J. Tamkun and C. Thummel for genomic and cDNA libraries; C. Bacot for sequencing; K. Womble for assistance with imaging, and B. Ring for help with formatting. This work was supported in part by a grant from the Florida chapter of the American Cancer Society and a Basil O'Connor Starter Scholar Award from the March of Dimes Foundation.
Abbreviations
- ABC
ATP-binding cassette
- NBD
nucleotide-binding domain
- TM
transmembrane domain
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession no. AF270979).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160271797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160271797
References
- 1.Riddiford L M. In: The Development of Drosophila melanogaster. Bate M, Arias A M, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 899–939. [Google Scholar]
- 2.Truman J W, Taylor B J, Awad T A. In: The Development of Drosophila melanogaster. Bate M, Arias A M, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 1245–1275. [Google Scholar]
- 3.Fristrom D, Fristrom J W. In: The Development of Drosophila melanogaster. Bate M, Arias A M, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 843–897. [Google Scholar]
- 4.Koelle M R, Talbot W S, Segraves W A, Bender M T, Cherbas P, Hogness D S. Cell. 1991;67:59–77. doi: 10.1016/0092-8674(91)90572-g. [DOI] [PubMed] [Google Scholar]
- 5.Yao T P, Forman B M, Jiang Z, Cherbas L, Chen J D, McKeown M, Cherbas P, Evans R M. Nature (London) 1993;366:476–479. doi: 10.1038/366476a0. [DOI] [PubMed] [Google Scholar]
- 6.Talbot W S, Swyryd E A, Hogness D S. Cell. 1993;73:1323–1337. doi: 10.1016/0092-8674(93)90359-x. [DOI] [PubMed] [Google Scholar]
- 7.Bender M, Imam F, Talbot W, Ganetzky B, Hogness D S. Cell. 1997;91:777–788. doi: 10.1016/s0092-8674(00)80466-3. [DOI] [PubMed] [Google Scholar]
- 8.Arbeitman M N, Hogness D S. Cell. 2000;101:67–77. doi: 10.1016/S0092-8674(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 9.White K P, Rifkin S A, Hurban P, Hogness D S. Science. 1999;286:2179–2184. doi: 10.1126/science.286.5447.2179. [DOI] [PubMed] [Google Scholar]
- 10.Ashburner M, Chihara C, Meltzer P, Richards G. Cold Spring Harbor Symp Quant Biol. 1974;38:655–662. doi: 10.1101/sqb.1974.038.01.070. [DOI] [PubMed] [Google Scholar]
- 11.Thummel C S. Trends Genet. 1996;12(8):306–310. doi: 10.1016/0168-9525(96)10032-9. [DOI] [PubMed] [Google Scholar]
- 12.Holland B, Blight M. J Mol Biol. 1999;293:381–399. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- 13.Ambudkar S, Dey S, Hrycyna C, Ramachandra M, Pastan I, Gottesman M. Annu Rev Pharmacol Toxicol. 1999;9:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 14.Young S, Fielding C. Nat Genet. 1999;22:316–318. doi: 10.1038/11878. [DOI] [PubMed] [Google Scholar]
- 15.Klein I, Sarkadi B, Varadi A. Biochim Biophys Acta. 1999;1461:237–262. doi: 10.1016/s0005-2736(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 16.Garza D, Ajioka J W, Burke D T, Hartl D L. Science. 1989;246:641–646. doi: 10.1126/science.2510296. [DOI] [PubMed] [Google Scholar]
- 17.Stowers R S, Russell S, Garza D. Dev Biol. 1999;213:116–130. doi: 10.1006/dbio.1999.9358. [DOI] [PubMed] [Google Scholar]
- 18.Andres A J, Thummel C S. Methods Cell Biol. 1994;44:565–573. doi: 10.1016/s0091-679x(08)60932-2. [DOI] [PubMed] [Google Scholar]
- 19.Karim F D, Thummel C S. Genes Dev. 1991;5:1067–1079. doi: 10.1101/gad.5.6.1067. [DOI] [PubMed] [Google Scholar]
- 20.Ashburner M. Chromosoma. 1972;38:255–281. doi: 10.1007/BF00290925. [DOI] [PubMed] [Google Scholar]
- 21.Rubin G M, Spradling A C. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 22.Higgins C. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 23.Segraves W A, Hogness D S. Genes Dev. 1990;4:204–219. doi: 10.1101/gad.4.2.204. [DOI] [PubMed] [Google Scholar]
- 24.Thummel C S, Burtis K C, Hogness D S. Cell. 1990;61:101–111. doi: 10.1016/0092-8674(90)90218-4. [DOI] [PubMed] [Google Scholar]
- 25.Huet F, Ruiz C, Richards G. Development (Cambridge, UK) 1993;118:613–627. doi: 10.1242/dev.118.2.613. [DOI] [PubMed] [Google Scholar]
- 26.Karim F D, Guild G M, Thummel C S. Development (Cambridge, UK) 1993;118:977–988. doi: 10.1242/dev.118.3.977. [DOI] [PubMed] [Google Scholar]
- 27.Adams M D, Celniker S E, Holt R A, Evans C A, Gocayne J D, Amanatides P G, Scherer S E, Li P W, Hoskins R A, Galle R F, et al. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 28.Arceci R J, Baas F, Raponi R, Horwitz S B, Housman D, Croop J M. Mol Reprod Dev. 1990;25:101–109. doi: 10.1002/mrd.1080250202. [DOI] [PubMed] [Google Scholar]
- 29.Altuvia S, Stein W D, Goldenberg S, Kane S E, Pastan I, Gottesman M M. J Biol Chem. 1993;268:27127–27132. [PubMed] [Google Scholar]
- 30.Bourgeois S, Gruol D J, Newby R F, Rajah R M. Mol Endocrinol. 1993;7:840–851. doi: 10.1210/mend.7.7.8105374. [DOI] [PubMed] [Google Scholar]
- 31.Venkateswaran A, Repa J, Lobaccaro J, A, B, Mangelsdorf D, Edwards P. J Biol Chem. 2000;275:14700–14707. doi: 10.1074/jbc.275.19.14700. [DOI] [PubMed] [Google Scholar]
- 32.Klucken J, Buchler C, Orso E, Kaminski W, Porsch-Orcurumez M, Liebisch G, Kapinsky M, Diederich W, Drobnik W, Dean M, et al. Proc Natl Acad Sci USA. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sliter T. Development (Cambridge, UK) 1989;106:347–354. doi: 10.1242/dev.106.2.347. [DOI] [PubMed] [Google Scholar]
- 34.Lam G T, Jiang C, Thummel C S. Development (Cambridge, UK) 1997;124:1757–1769. doi: 10.1242/dev.124.9.1757. [DOI] [PubMed] [Google Scholar]
- 35.White K P, Hurban P, Watanabe T, Hogness D S. Science. 1997;276:114–117. doi: 10.1126/science.276.5309.114. [DOI] [PubMed] [Google Scholar]
- 36.Lam G, Hall B L, Bender M, Thummel C S. Dev Biol. 1999;212:204–216. doi: 10.1006/dbio.1999.9343. [DOI] [PubMed] [Google Scholar]
- 37.Kralli A, Bohen S P, Yamamoto K R. Proc Natl Acad Sci USA. 1995;92:4701–4705. doi: 10.1073/pnas.92.10.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahe Y, Lemoine Y, Kuchler K. J Biol Chem. 1996;217:25167–25172. doi: 10.1074/jbc.271.41.25167. [DOI] [PubMed] [Google Scholar]
- 39.von Kalken C K, Broxterman H J, Pinedo H M, Feller N, Dekker H, Lankelma J, Giaccone G. Br J Cancer. 1993;67:284–289. doi: 10.1038/bjc.1993.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gruol D, Bourgeois S. Biochem Cell Biol. 1994;72:561–571. doi: 10.1139/o94-075. [DOI] [PubMed] [Google Scholar]
- 41.Gruol D, Vo Q, Zee M. Biochem Pharmacol. 1999;58:1191–1199. doi: 10.1016/s0006-2952(99)00201-4. [DOI] [PubMed] [Google Scholar]
- 42.Wu U C, Horvitz H. Cell. 1998;93:951–960. doi: 10.1016/s0092-8674(00)81201-5. [DOI] [PubMed] [Google Scholar]
- 43.Johnstone R, Ruefli A, Smyth M. Trends Biochem Sci. 2000;25:1–6. doi: 10.1016/s0968-0004(99)01493-0. [DOI] [PubMed] [Google Scholar]
- 44.Jiang C, Lamblin A-F J, Steller H, Thummel C S. Mol Cell. 2000;5:445–455. doi: 10.1016/s1097-2765(00)80439-6. [DOI] [PubMed] [Google Scholar]
- 45.Karim F D, Thummel C S. EMBO J. 1992;11:4083–4093. doi: 10.1002/j.1460-2075.1992.tb05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burtis K C, Thummel C S, Jones C W, Karim F D, Hogness D S. Cell. 1990;61:85–99. doi: 10.1016/0092-8674(90)90217-3. [DOI] [PubMed] [Google Scholar]
- 47.Richards G. Dev Biol. 1976;54:264–275. doi: 10.1016/0012-1606(76)90304-3. [DOI] [PubMed] [Google Scholar]
- 48.Broadus J, McCabe J, Endrizzi B, Thummel C S, Woodard C T. Mol Cell. 1999;3:143–149. doi: 10.1016/s1097-2765(00)80305-6. [DOI] [PubMed] [Google Scholar]
- 49.Richards G. Biol Rev. 1981;56:501–549. [Google Scholar]
- 50.Koolman J, Karlson P. In: Comprehensive Insect Physiology, Biochemistry and Pharmacology. Kerkut G, Gilbert L, editors. Vol. 7. Oxford: Pergamon; 1985. pp. 343–390. [Google Scholar]