

Abstract
The pathomechanism of familial hypokalemic periodic paralysis (HypoPP) is a mystery, despite knowledge of the underlying dominant point mutations in the dihydropyridine receptor (DHPR) voltage sensor. In five HypoPP families without DHPR gene defects, we identified two mutations, Arg-672→His and →Gly, in the voltage sensor of domain 2 of a different protein: the skeletal muscle sodium channel α subunit, known to be responsible for hereditary muscle diseases associated with myotonia. Excised skeletal muscle fibers from a patient heterozygous for Arg-672→Gly displayed depolarization and weakness in low-potassium extracellular solution. Slowing and smaller size of action potentials were suggestive of excitability of the wild-type channel population only. Heterologous expression of the two sodium channel mutations revealed a 10-mV left shift of the steady-state fast inactivation curve enhancing inactivation and a sodium current density that was reduced even at potentials at which inactivation was removed. Decreased current and small action potentials suggested a low channel protein density. The alterations are decisive for the pathogenesis of episodic muscle weakness by reducing the number of excitable sodium channels particularly at sustained membrane depolarization. The results prove that SCN4A, the gene encoding the sodium channel α subunit of skeletal muscle is responsible for HypoPP-2 which does not differ clinically from DHPR-HypoPP. HypoPP-2 represents a disease caused by enhanced channel inactivation and current reduction showing no myotonia.
Keywords: channelopathy, molecular genetics, functional expression, muscle disease
Two dominantly inherited clinically similar types of episodic flaccid generalized weakness are known: hypokalemic periodic paralysis (HypoPP) and hyperkalemic periodic paralysis (HyperPP), distinguished by the changes in serum potassium levels during paralytic attacks. An important clinical difference between the two entities is triggers of attacks of weakness, e.g., HyperPP can be provoked by oral potassium administration, whereas this is a remedy for HypoPP. Cooccurrence of myotonia is found in HyperPP but not in HypoPP patients (1).
Genetic studies revealed that point mutations in the gene encoding the α subunit of the adult skeletal muscle sodium channel (SCN4A) are causative for HyperPP (2–4). This gene encodes the main pore-forming subunit, which consists of four domains of internal homology (D1–D4), each containing six putative transmembrane segments (S1–S6), of which the S4 segments are thought to function as the voltage sensor for channel gating. Pathogenetically, the long-lasting muscle fiber membrane depolarization leading to weakness in HyperPP patients is caused by incomplete sodium channel inactivation and persistent current of the mutant (5, 6). To date, five mutations have been described, none of which are directly located in the voltage sensors.
In contrast, the causative gene for HypoPP, CACNA1S, was shown to encode the α1 subunit of the dihydropyridine (DHP) receptor (7, 8), the pore-forming subunit of the pentameric DHP-sensitive calcium channel in the transverse tubular membrane that is thought to interact with the intracellular calcium release channel in mediation of excitation-contraction coupling. Three mutations localized in D2/S4 and D4/S4 have been described, but their effects on channel function have not been clarified. Changes in activation, inactivation, and secondary effects all have been suggested to relate to disease pathogenesis (9–13).
Very recently, a point mutation, Arg-669→His, of D2/S4 of the SCN4A gene, was reported in a single HypoPP family of four affected individuals (14). To test SCN4A as causative for the disease in several typical HypoPP families for which DHPR gene defects were excluded (15), we performed genetic linkage studies and screened for mutations. We found two SCN4A base exchanges and identified them as disease-causing mutations by functional expression studies.
Materials and Methods
Families.
Members of all five families participating in the study gave their informed consent. Experiments were approved by the Ethics Committee of Ulm University and Groupe Hospitalier Pitié-Salpêtrière and were in concordance with the Declaration of Helsinki. All affected individuals except one were personally examined by one of us.
In four (HypoPP106,6,29,105) of the five pedigrees studied, an autosomal dominant mode of inheritance of episodic weakness was evident (Figs. 1 and 2). One pedigree had only one clinically affected individual (HypoPP18). The families were chronologically numbered to permit comparison with earlier reports (16, 17). Decisive criteria used for diagnosis of HypoPP were: hypokalemia between 1 mM and 2.7 mM during the attack in at least one individual of each family, amelioration of symptoms by oral potassium in at least one individual, absence of myotonia in the electromyogram in all family members, and presence of tubular aggregates in muscle biopsy in one patient of each kinship.
Figure 1.
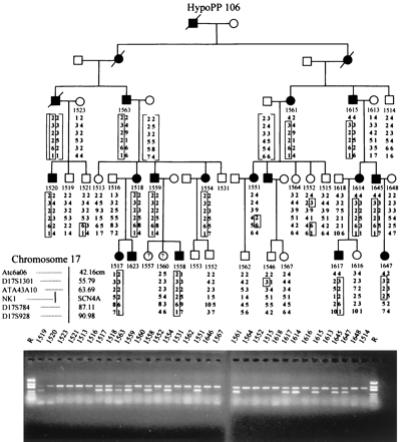
Pedigree of family HypoPP106 and genetic map of the SCN4A locus on chromosome 17q. Patients are represented by filled symbols and unaffected individuals by open symbols (square = males; circles = females). The haplotype segregating with the disease is boxed. A marker map is on the left below the pedigree. (Lower) A polyacrylamide gel electrophoresis of the C-2015-G allele-specific PCR product (265 bp) shows cosegregation of the mutation encoding Arg-672→Gly without recombinants. The additional constant band of 386 bp is the PCR product of SCN4A exon 6 in the same reaction mix, demonstrating DNA integrity of each sample.
Figure 2.
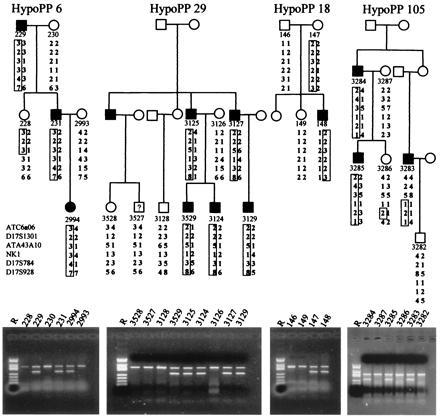
Pedigrees of families HypoPP6, HypoPP29, HypoPP18, and HypoPP105. Symbols and markers are as in Fig. 1. In addition to the allele-specific marker shown for C-2015-G for HypoPP6, an allele-specific marker for the G-2016-A transition in exon 12 predicting Arg-672→His was used for families HypoPP29, HypoPP18, and HypoPP105 of 266 bp. The control band of 386 bp was SCN4A exon 6 in all cases.
Genetics.
Linkage analysis for exclusion of linkage to chromosome 1 (CACNA1S locus) and inclusion of chromosome 17 (SCN4A locus) was performed by using Généthon microsatellite markers, as previously described (18). For at least one patient of each family, all three known CACNA1S mutations were excluded by restriction analysis and single-strand conformation analysis (SSCA), as described previously (7). Linkage to the CACNA1S locus on chromosome 1q32 was excluded for HypoPP106 (15) and HypoPP29 (data not shown). For the other pedigrees, linkage data were not informative. Logarithm of odds scores were calculated by using mlink of the linkage package (Jurg Ott, Zurich) with an assumed penetrance of 98% and an allele frequency of 1:100,000. Mutation analysis from genomic DNA using the SSCA technique was performed in the SCN4A gene in all exons encoding the transmembrane segments near the pore of the channel (exons 6, 7, 11–14, 20, 24). Aberrant bands were sequenced by using an automated 373A sequencer (Applied Biosystems). An allele-specific PCR test was developed to test for the SCN4A mutations identified.
Recordings from Native Muscle Fiber Segments.
Muscle specimens were removed, under local or regional anesthesia, from the male index patient of family HypoPP6 (Arg-672→Gly) and five adult individuals with no neuromuscular disease. The specimens were about 3 cm in length and 0.5 cm in diameter. All measurements were performed in a standard solution maintained at 37°C and containing (in mM): NaCl, 108; KCl, 5.0; CaCl2, 1.5; MgSO4, 0.7; NaHCO3, 26.2; NaH2PO4, 1.7; Na gluconate, 9.6; glucose, 5.5; and sucrose, 7.6; pH was adjusted to 7.4 by gassing with 95% O2 and 5% CO2. The KATP channel activator cromakalim (BRL 34915; Beecham Pharmaceuticals) was prepared in DMSO (Sigma).
Isometric contractions of muscle bundles 2–3 mm in diameter were induced by supramaximal field stimulation at a frequency of 0.1 Hz and recorded by using a force transducer (Grass Instruments, Quincy, MA, FT03). To determine specific membrane conductance and capacitance, single-fiber segments were impaled midway between the ends with three capacity-compensated microelectrodes, as previously described (5, 17). Action potentials were elicited by brief depolarizing current pulses superposing a constant current of variable amplitude, which produced holding potentials between −55 and −120 mV. Action potentials were recorded by an intracellular microelectrode in the vicinity of the endplate. As the maximum rate of rise of an action potential depends on the maximum sodium conductance (19), its dependence on the holding potential yielded a steady-state inactivation curve describable by the Boltzmann equation: V′/V′max = 1/(1 + exp(V − Vh0.5)/k). Action potential duration was defined from the upstroke to 90% of complete repolarization. Pooled data are represented as means and standard deviations. Statistical differences were evaluated by Student's t test.
Mutagenesis and Patch-Clamp Experiments.
Site-directed mutagenesis was performed by using an overlapping technique PCR-based technique. Subsequently, the mutants were reassembled in the pRC/CMV plasmid (Invitrogen) for transfection by the calcium phosphate precipitation method in tsA201, a mammalian cell line. No auxiliary β subunit was coexpressed, as it binds to a region very different from D2/S4, and its effect is small in a mammalian cell in contrast to expression in Xenopus oocytes (4).
Standard whole-cell recording methods were used, as previously described (20, 21). The pulse protocols are given in Results and the figure legends. Capacity transients were eliminated by -P/4 protocol. Series resistance errors were <3 mV. Data were filtered at 3–10 kHz and acquired by using pclamp (Axon Instruments, Foster City, CA). Patch electrodes contained (in mM): 105 CsF, 35 NaCl, 10 EGTA, 10 Cs-Hepes, pH 7.4. The bath contained 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 Cs-Hepes, pH 7.4. Corrections were made for liquid junction potentials. Most experiments were done at room temperature (20–22°C). In a few experiments, the temperature was set to 15°C by use of a feedback-regulated Peltier device (Peschel, Gummersbach, Germany). Whole-cell data were displayed and analyzed by a combination of pclamp and origin (Microcal Software, Northampton, MA) programs. Data are presented as means and standard errors of the estimated fit parameters.
Results
Families and Genetics.
Linkage testing to the SCN4A locus on chromosome 17q was informative only for HypoPP106. The following maximal two-point logarithm of odds scores for the three markers flanking the region were obtained: 2.27 for ATA43A10 (at a recombination frequency of Θ = 0.07), 6.65 for the intragenic marker NK1 (Θ = 0), and 1.67 for D17S784 (Θ = 0.15) (for haplotypes of all pedigrees, see Figs. 1 and 2).
SSCA screening yielded aberrant bands in exon 12 in all 5 unrelated screening individuals. Sequencing revealed a C-2015-G base exchange encoding an Arg-672→Gly substitution in two samples (HypoPP6 and 106) and a G-2016-A transition in three additional samples coding for amino acid variation Arg-672→His. Arg-672 is the second outermost arginine of the helical transmembrane segment D2/S4 that participates in voltage sensing. This arginine is highly conserved in all voltage-gated sodium channel α subunits and moreover, in all calcium channel α1 subunits sequenced to date (GenBank accession no):
Human skeletal: 661 NVQGLSVLRSFRLLRVFKLAKSWPTLN(NM_000334.1)
Human brain:842 NVEGLSVLRSFRLLRVFKLAKSWPTLN (Q99250)
Human cardiac:588 NVAGMALLRLFRMLRIFKLGKYWPTFQ (NM_002976.1)
Rat skeletal:655NVQGLSVLRSFRLLRVFKLAKSWPTLN (P15390)
Fugu skeletal:683NVQGLSVLRSFRLLRVFKLAKSWPTLN (AB030482.1)
The allele-specific PCR for the mutations revealed no further incidence of the base exchanges in 60 sporadic HypoPP cases without a calcium channel mutation, 80 mainly sporadic HyperPP patients without a known HyperPP mutation, and 70 healthy controls. In our five families, the product was obtained only in carriers of the base exchanges cosegregating without recombinants with the disease phenotype in four families. The unaffected mother of the patient in family HypoPP18 was shown to be an obligate carrier, suggesting nonpenetrance in females that has been also described for the calcium channel variant of the disease (22) (Figs. 1 and 2 Lower).
Native Muscle Fiber Segments. Force measurements.
After a delay of about 10 minutes, lowering of external potassium concentration to 1 mM led to a reproducible reduction of twitch force of the three HypoPP6 muscle bundles, to a mean of 53% of the strength yielded in 5 mM potassium solution, in agreement with the diagnosis of HypoPP (16). Addition of 100 μM cromakalim, a KATP channel activator, to the low-potassium solution increased muscle strength, mostly beyond the original amplitude, indicative of partial paralysis of the bundles. Also typical for HypoPP and in contrast to HyperPP muscle, 100 units per liter of insulin and 1 μM of adrenaline in the bathing solution further reduced the force of two bundles to 42% and 35% of the initial value, respectively. Different from most other HypoPP muscle samples (16), all bundles regained force after washout of the provocative factors.
Resting membrane potentials.
In a bathing solution containing 5 mM potassium, the resealed fiber segments of patient HypoPP6 were slightly depolarized to −66 ± 9 mV (n = 47 fibers) compared to normal resting values of −74 ± 10 mV (n = 52). The absence of statistical significance may be caused by the use of cut fibers that show a greater potential variability regarding resealing and repolarization than the intact intercostal fibers of a previous study on HypoPP (17). Exposure to 1 μM tetrodotoxin did not significantly change the resting potentials. Similar to ref. 17, exposure of the HypoPP6 fiber segments to 1 mM potassium solution reproducibly induced further depolarization to −55 ± 9 mV (n = 8; P < 0.05), whereas the majority of muscle fiber segments from normal controls responded with hyperpolarization.
Specific membrane conductance and capacitance.
There was no difference between the steady-state current density-voltage relationships derived from the HypoPP6 patient and normal controls. The slopes of the relationships, which at −80 mV correspond to the total conductance of the resting membrane, were determined as 307 ± 102 μs/cm2 (n = 9) for HypoPP6 and 271 ± 43 μs/cm2 (n = 10; P > 0.05) for healthy individuals. The unaltered value indicates that the major resting ion conductances in HypoPP6 were unchanged compared to controls. The specific membrane capacitance for HypoPP 6 (3.9 ± 0.7 μF/cm2, n = 7) also did not differ from that of controls (3.4 ± 0.6 μF/cm2, n = 17; P > 0.5).
Action Potentials.
For all holding potentials. HypoPP6 action potentials revealed lower maximum rates of rise and smaller peak potentials. When elicited from −80 mV, rate of rise was 175 ± 46 V/s (n = 8) compared to 370 ± 87 V/s (n = 30; P < 0.001) for controls; HypoPP6 peak potential was −9.1 ± 8.0 mV (n = 8) vs. +10.5 ± 7.9 mV (n = 37); P < 0.05; Fig. 4D). Hyperpolarization to −120 mV increased maximum rates of rise and peak potentials only slightly. The action potentials lasted 2.41 ± 1.20 ms (n = 13) in contrast to the normal duration of 1.32 ± 0.95 ms (n = 21; P < 0.01). These abnormal values were independent of the initial resting potential as the holding potential of −80 mV at the recording site was kept stable by a constant current for more than 3 minutes before the first depolarization pulse. Analysis of the maximum rate of rise revealed that the absolute peak rates for HypoPP6 fibers were approximately 50% of the control values for each holding potential between −55 and −100 mV. The voltage dependence of the normalized peak rates, i.e., the steady-state inactivation curve, was therefore almost identical for HypoPP and control fibers, showing a midpoint voltage of the Boltzmann fit, Vh0.5, at −65 mV and a slope factor, k, of 7.5 mV (n = 8 and 22).
Figure 4.
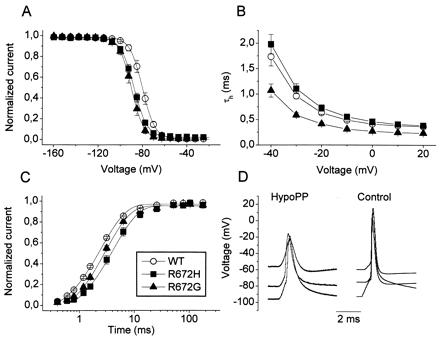
(A) Steady-state inactivation was determined from a holding potential of −160 mV by using a series of 300-ms prepulses from −160 to +27.5 mV in 7.5-mV increments before the test pulse to −20 mV. Note that all measurements were performed in chloride-reduced pipette solution containing fluoride and yielding highly negative Vh0.5. (B) Voltage dependence of time constants of fast inactivation time constants, τh, is given for both mutants and WT (n = 6–9). (C) Recovery from inactivation for a holding potential of −120 mV was determined by a 100-ms depolarization to −20 mV followed by a variable-duration return to −120 mV. (D) Representative action potentials from a muscle fiber segment of the HypoPP6 patient compared to those of a normal control. They were elicited from various holding potentials by a short depolarizing pulse. Note the slower rise and fall for HypoPP.
Conclusions from Experiments on Native HypoPP Fiber Segments.
According to mathematical models, a markedly reduced rate of rise of the action potentials should result in slowing of the propagation velocity of muscle fiber action potentials if other parameters, like membrane capacitance, are unaltered (23). A reduced velocity has been reported for HypoPP patients (24), supporting our unexpected findings. This reduced rate of rise to 50% could be explained easily by inexcitability of the mutant channels, at least in the available muscle samples that had initially depolarized during the biopsy procedure and incompletely recovered thereafter. The remaining wild-type (WT) sodium channels (50% of the population at unaltered expression of mutant channels) may be fully responsible for the normal potential dependence of steady-state inactivation of the available sodium channels and the generation of aborted action potentials. Considering the parallel effects of low extracellular potassium on the resting potential and twitch force, sustained membrane depolarization may be responsible for the reduction of muscle strength, probably by inactivation of normal sodium channels, causing inability of muscle fibers to generate action potentials that can be propagated. To better understand the functional alteration of the mutant sodium channels, whole-cell patch-clamp experiments on heterologously expressed channels were performed.
Functional Expression of Mutant Channels: Activation/Deactivation.
Fig. 3A shows typical sodium currents recorded from tsA-201 cells expressing mutant and WT α subunit channels. In general, the mutant channels did not express as well as the WT channels. The current density was significantly reduced for Arg-672→His (148 ± 59 p/pF; P < 0.01; n = 7) and for Arg-672→Gly (240 ± 45 pA/pF; P < 0.02; n = 7) in comparison to WT channels (583 ± 127 pA/pF; n = 5). Normalized peak current-voltage relationships were similar for the three channel populations tested (Fig. 3B). The Boltzmann fit parameters for normalized conductance-voltage relationships (not shown) revealed similar values for the potentials of half activation, Vm0.5 of −36 ± 1 (WT) vs. −39 ± 1 (His) vs. −35 ± 1 (Gly), but slightly increased slope factors, k = 8.0 ± 0.4 (His) vs. 7.9 ± 0.4 (Gly) compared to 6.7 ± 0.3 for WT (P < 0.05, n = 6–9). As the slope factor is inversely related to the steepness of the voltage dependence, the mutations produced a slightly less steep curve equivalent to a reduction of 0.6 elementary charges (e0). This is in agreement with the neutralization of the positively charged arginine by glycine or histidine in the domain D2 voltage sensor of the sodium channel (21, 25).
Figure 3.
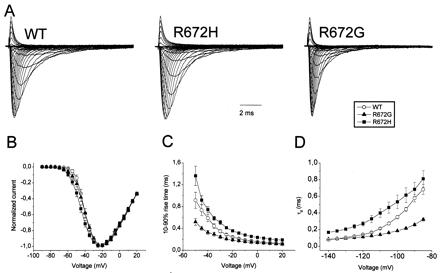
Sodium currents elicited by a family of 10-ms lasting depolarizations from a −140 mV holding potential to voltages ranging from −90 to +20 mV in 5-mV steps were recorded from tsA-201 cells expressing WT, Arg-672→His, and Arg-672→Gly α subunit channels. The maximum peaks were normalized to identical amplitudes. (B) Corresponding peak current-voltage relationships (n = 5–7) normalized to maximum (=100%). (C) Voltage dependence of the activation time constants is plotted against voltage steps. The kinetics of activation were estimated by determining the rise time between 10% and 90% (n = 5–9). (D) Deactivation kinetics measured at 15°C. Cells held at −140 mV were depolarized for 500 μs to +20 mV before stepping back to test potentials shown (n = 3–5).
The kinetics of activation were estimated by determining the 10–90% rise time to the maximal peak current. Arg-672→His showed a slower rise to the maximal peak (Fig. 3C), meaning slower kinetics of activation for the whole voltage range measured. In contrast, Arg-672→Gly showed accelerated activation kinetics.
Deactivation kinetics were measured at 15°C for better time resolution (Fig. 3D). Deactivation time constants were increased for Arg-672→His when compared to WT. For Arg-672→Gly, the deactivation was faster as in WT, in the range between −110 and −80 mV. At very negative potentials, the kinetics of deactivation revealed no difference. It is important to note that at these potentials, we are on the limit of resolution and may not be able to detect a possible faster deactivation of Arg-672→Gly vs. WT.
Inactivation and Recovery from Inactivation.
Steady-state inactivation was determined from a holding potential of −160 mV by using a series of 300-ms prepulses to the potentials before the test pulse to −20 mV (Fig. 4A). Boltzmann fit parameters yielded Vh0.5 (mV) of −80 ± 1 (WT), −88 ± 1 (Arg-672→His), −90 ± 2 (Arg-672→Gly) and k = 5.0 ± 0.1, 5.6 ± 0.3, 5.3 ± 0.3 (P > 0.05; n = 6–9). The significant 10-mV left shift of the steady-state inactivation curve found for both mutants shows that the number of available sodium channels is reduced.
We determined the fast inactivation time constant, τh from a monoexponential fit to the first 8 ms of the current decay of mutant and WT channels. τh was slightly increased for Arg-672→His (e.g., at −20 mV: 0.73 ± 0.06 ms) and decreased for Arg-672→Gly (0.41 ± 0.03 ms) when compared to WT (0.63 ± 0.03 ms) (Fig. 4B).
The size of the persistent current known to be important for resting potential and HyperPP pathogenesis (5, 26) was determined 50 ms after the beginning of the depolarizing pulse. For both hypoPP mutants, the persistent current was not increased when compared to WT (<0.5% of the peak current; n = 6 each).
Recovery from inactivation was determined for a holding potential of −120 mV. Channels were inactivated by a 100-ms depolarization of −20 mV from the holding potential. Subsequently, a variable-duration return to −120 mV induced recovery from inactivation. Both mutants showed slower recovery from inactivation (Fig. 4C). For Arg-672→His, recovery was 1.5-fold slower when compared to WT channels (4.00 ± 0.35 ms vs. 2.68 ± 0.06 ms, n = 5–6; P < 0.01). Recovery from inactivation was also significantly slower for Arg-672→Gly (3.20 ± 0.14 ms, P < 0.01, n = 4), indicating stabilization of the fast inactivated state, thus in accordance with the left shift of steady-state inactivation.
The effects of the mutations on slow inactivation were studied by the following steady-state inactivation protocol: slow inactivation was induced by a series of 30-s depolarizing prepulses ranging from −140 to +20 mV. Fast inactivation was removed by a 20-ms pulse to −140 mV before the test pulse to −20 mV. Both mutations changed the properties of slow inactivation. The R672G mutation showed a 10-mV left shift of the steady-state inactivation (Vh0.5 = −78 ± 2, k = 12.2 ± 0.8, n = 4) vs. WT (Vh0.5 = −68 ± 2, k = 11.3 ± 0.4, n = 11), clearly stabilizing the slow inactivated state. However, this histidine substitution for the R672 showed a 7-mV right shift of the steady-state inactivation and a reduction of the voltage dependence (Vh0.5 = −61 ± 1, k = 19.2 ± 1.2, n = 6). The opposite effects of the mutants suggest that the effects on slow inactivation may not be the decisive ones for disease pathogenesis.
Discussion
Typical clinical features in the five families of the study and in vitro loss of muscle fiber strength at low extracellular potassium associated with membrane depolarization on a muscle specimen of one of the patients ensure the diagnosis of HypoPP. The genetic data presented here strongly suggest the SCN4A gene to be a second gene for HypoPP: perfect segregation of the base exchanges with the phenotype, no occurrence in healthy controls, and predicted amino acid exchanges in a highly conserved region of functional importance, i.e., the voltage sensor of the channel. Finally, demonstration of functional changes brought about by the base exchanges proves that they are disease-causing mutations and that SCN4A is the gene responsible for HypoPP-2, a form of HypoPP clinically indistinguishable from DHPR-related HypoPP, i.e., HypoPP-1.
Functionally, slowing of sodium channel kinetics of activation, inactivation, and deactivation for Arg-672→His suggests that the histidine residue hinders S4 movement regardless of direction. In contrast, glycine, the smallest possible residue, seems to accelerate movement of the S4 segment compared to both histidine and WT arginine. Because of the opposite effects of the two mutations, we considered these alterations of minor importance for the pathomechanism of HypoPP.
Two pathogenetically decisive alterations could be the left shift of the steady-state inactivation curve of the mutant channel and the reduced sodium current density. Both alterations have the same effect, hypoexcitability of the fiber membrane resulting in muscle weakness; (i) the left shift by 10 mV reduces the number of excitable mutant sodium channels already at the normal resting potential of approximately −80 mV; any sustained membrane depolarization because of mechanisms discussed below will further decrease the number of excitable sodium channels and make the generation and propagation of action potentials impossible; (ii) in addition to the current reduction because of the left shift, a decreased sodium current density in the expression system, as well as a reduction of action potential size of native HypoPP fibers, was measured. Both alterations were found at highly negative holding potentials at which inactivation is removed, pointing to a reduced channel open probability or reduced channel protein density; the latter could be explained by impaired expression of the mutant gene, by RNA/protein instabilities, or by disturbed trafficking to or insertion into the membrane.
In HypoPP, hypokalemia results from the well-known physiological effect of glucose intake and the release of insulin, which stimulates the sodium-potassium pump and shifts potassium ions from the extracellular space into the intracellular compartment (27). In contrast to the membrane hyperpolarization observed in normal muscle, the hypokalemia causes sustained depolarization of HypoPP fibers and initiates the attack (17, 28). This hypokalemia-induced depolarization has been shown to be associated with an inward rectifier potassium current, the outward component of which is apparently blocked by low-potassium and insulin in HypoPP (28, 29). In HypoPP-2, the sustained depolarization will cause further inactivation of the mutant sodium channels and, when progressing, will inactivate more and more normal sodium channels and render the cell inexcitable, i.e., the patient will be paralyzed. Such secondary effects, like changes of potassium channel expression patterns or proliferation of the transverse tubular system that leads to the tubular aggregate myopathy in HypoPP (30), are also found in other myopathies; e.g., in myotonic dystrophy, hyperexcitability is associated with overexpression of an apamin-sensitive potassium channel (31). Expression profiling by the use of DNA chip techniques will soon enable better understanding of myopathy-related mechanisms.
The question of how the SCN4A mutations could produce such a specific clinical phenotype as HypoPP may not be fully answered, though. All three known mutations affect D2/S4 arginines, and no nonsense mutation was identified despite screening 30% of the total coding sequence. If complete loss of function were the mechanism of pathogenesis, a certain amount of null mutations would be expected, as is the case for episodic ataxia type 2 or benign neonatal convulsions caused by loss-of-function mutations in neuronal calcium and potassium channels, respectively (32, 33). Perhaps a total loss of function causes a more severe phenotype with not necessarily episodic symptoms. Therefore, a specific change of function under special conditions not touched by our experiments is possible.
Among designed mutations in the various S4 voltage sensors of a brain sodium channel, only those in D2 caused an isolated and large left shift of the steady-state fast and slow inactivation curve (34). The closed-state inactivation is also enhanced by a β-scorpion toxin that requires a depolarization-induced movement of D2/S4 for binding (35). Both reports underline the specific role of D2/S4, which, in contrast to D4/S4, is not involved in activation-inactivation coupling (36). All human sodium channel mutations, including those in D4/S4 previously functionally studied, showed gain of function because of destabilized inactivation. All are associated with myotonia, a symptom never occurring in HypoPP (1). Only a few of these mutations additionally caused a left shift of fast inactivation and these were regularly associated with episodic muscle weakness in carriers (37, 38). None of the myotonia-causing mutations are localized in D2/S4, supporting our hypothesis that enhanced inactivation contributes to periodic paralysis, but that it may not be the only mechanism.
Acknowledgments
We thank Drs. R. Rüdel and W. Melzer for fruitful discussions and S. Scherr, U. Pika-Hartlaub, and U. Richter for technical assistance. This work was a project of the Interdisciplinary Clinical Research Center Ulm, additionally supported by Deutsche Forschungsgemeinschaft (Le 481/4-1, Mi 472/5-1), the European Community, Association Française contre les Myopathies, and Institut National de la Santé et de la Recherche Médicale.
Abbreviations
- HypoPP
hypokalemic periodic paralysis
- HyperPP
hyperkalemic periodic paralysis
- SCN4A
α subunit of the adult skeletal muscle sodium channel
- DHP
dihydropyridine
- SSCA
single-strand conformation analysis
- WT
wild type
References
- 1.Lehmann-Horn F, Engel A, Rüdel R, Ricker K. In: Myology. Engel A G, Franzini-Armstrong C, editors. New York: McGraw-Hill; 1994. pp. 1304–1334. [Google Scholar]
- 2.Fontaine B, Khurana T S, Hoffman E P, Bruns G A, Haines J L, Trofatter J A, Hanson M P, Rich J, McFarlane H, McKenna-Yasek D, et al. Science. 1990;250:1000–1003. doi: 10.1126/science.2173143. [DOI] [PubMed] [Google Scholar]
- 3.Rojas C V, Wang J Z, Schwartz L S, Hoffman E P, Powell B R, Brown R H., Jr Nature (London) 1991;354:387–389. doi: 10.1038/354387a0. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann-Horn F, Jurkat-Rott K. Physiol Rev. 1999;79:1317–1371. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann-Horn F, Küther G, Ricker K, Grafe P, Ballanyi K, Rüdel R. Muscle Nerve. 1987;10:363–374. doi: 10.1002/mus.880100414. [DOI] [PubMed] [Google Scholar]
- 6.Cummins T R, Sigworth F J. Biophys J. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg R G, Hogan K, Powers P A, Lapie P, Vale-Santos J E, Weissenbach J, et al. Hum Mol Genet. 1994;3:1415–1419. doi: 10.1093/hmg/3.8.1415. [DOI] [PubMed] [Google Scholar]
- 8.Ptacek L J, Tawil R, Griggs R C, Engel A G, Layzer R B, Kwiecinski H, McManis P G, Santiago L, Moore M, Fouad G, et al. Cell. 1994;77:863–868. doi: 10.1016/0092-8674(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann-Horn F, Sipos I, Jurkat-Rott K, Heine R, Brinkmeier H, Fontaine B, Kovacs L, Melzer W. Soc Gen Physiol Ser. 1995;50:101–113. [PubMed] [Google Scholar]
- 10.Lapie P, Goudet C, Nargeot J, Fontaine B, Lory P. FEBS Lett. 1996;382:244–248. doi: 10.1016/0014-5793(96)00173-1. [DOI] [PubMed] [Google Scholar]
- 11.Jurkat-Rott K, Uetz U, Pika-Hartlaub U, Powell J, Fontaine B, Melzer W, Lehmann-Horn F. FEBS Lett. 1998;423:198–204. doi: 10.1016/s0014-5793(98)00090-8. [DOI] [PubMed] [Google Scholar]
- 12.Morrill J A, Brown R H, Jr, Cannon S C. J Neurosci. 1998;18:10320–10334. doi: 10.1523/JNEUROSCI.18-24-10320.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrill J A, Cannon S C. J Physiol (London) 1999;520:321–336. doi: 10.1111/j.1469-7793.1999.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulman D E, Scoggan K A, van Oene M D, Nicolle M W, Hahn A F, Tollar L L, Ebers G C. Neurology. 1999;53:1932–1936. doi: 10.1212/wnl.53.9.1932. [DOI] [PubMed] [Google Scholar]
- 15.Plassart E, Elbaz A, Santos J V, Reboul J, Lapie P, Chauveau D, Jurkat-Rott K, Guimaraes J, Saudubray J M, Weissenbach J, et al. Hum Genet. 1994;94:551–556. doi: 10.1007/BF00211025. [DOI] [PubMed] [Google Scholar]
- 16.Iaizzo P A, Quasthoff S, Lehmann-Horn F. Neuromuscul Disord. 1995;5:115–124. doi: 10.1016/0960-8966(94)00036-9. [DOI] [PubMed] [Google Scholar]
- 17.Rüdel R, Lehmann-Horn F, Ricker K, Küther G. Muscle Nerve. 1994;7:110–120. doi: 10.1002/mus.880070205. [DOI] [PubMed] [Google Scholar]
- 18.Fontaine B, Vale-Santos J, Jurkat-Rott K, Reboul J, Plassart E, Rime C S, Elbaz A, Heine R, Guimaraes J, Weissenbach J, et al. Nat Genet. 1994;6:267–272. doi: 10.1038/ng0394-267. [DOI] [PubMed] [Google Scholar]
- 19.Adrian R H, Peachey L D. J Physiol (London) 1973;235:103–131. doi: 10.1113/jphysiol.1973.sp010380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 21.Mitrovic N, George A L, Jr, Horn R. J Gen Physiol. 1998;111:451–462. doi: 10.1085/jgp.111.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elbaz A, Vale-Santos J, Jurkat-Rott K, Lapie P, Ophoff R A, Bady B, Links T P, Piussan C, Vila A, Monnier N, et al. Am J Hum Genet. 1995;56:374–380. [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter P J, McNaughton P A, Noble D. Prog Biophys Mol Biol. 1975;30:99–144. doi: 10.1016/0079-6107(76)90007-9. [DOI] [PubMed] [Google Scholar]
- 24.Troni W, Doriguzzi C, Mongini T. Neurology. 1983;33:1522–1525. doi: 10.1212/wnl.33.11.1522. [DOI] [PubMed] [Google Scholar]
- 25.Stühmer W, Conti F, Suzuki H, Wang X D, Noda M, Yahagi N, Kubo H, Numa S. Nature (London) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 26.Cannon S C, Brown R H, Jr, Corey D P. Neuron. 1991;6:619–626. doi: 10.1016/0896-6273(91)90064-7. [DOI] [PubMed] [Google Scholar]
- 27.Minaker K L, Meneilly G S, Flier J S, Rowe J W. Am J Med. 1988;84:1001–1006. doi: 10.1016/0002-9343(88)90304-x. [DOI] [PubMed] [Google Scholar]
- 28.Ruff R L. Neurology. 1999;53:1556–1563. doi: 10.1212/wnl.53.7.1556. [DOI] [PubMed] [Google Scholar]
- 29.Tricarico D, Servidei S, Tonali P, Jurkat-Rott K, Camerino D C. J Clin Invest. 1999;103:675–682. doi: 10.1172/JCI4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel A G. Mayo Clin Proc. 1970;45:774–814. [PubMed] [Google Scholar]
- 31.Renaud J F, Desnuelle C, Schmid-Antomarchi H, Hugues M, Serratrice G, Lazdunski M. Nature. 1986;319:678–680. doi: 10.1038/319678a0. [DOI] [PubMed] [Google Scholar]
- 32.Ophoff R A, Terwindt G M, Vergouwe M N, van Eijk R, Oefner P J, Hoffman S M G, Lamerdin J E, Mohrenweiser H W, Bulman D E, Ferrari M, et al. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 33.Biervert C, Schroeder B C, Kubisch C, Berkovic S F, Propping P, Jentsch T J, Steinlein O K. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 34.Kontis K J, Goldin A L. J Gen Physiol. 1997;110:403–413. doi: 10.1085/jgp.110.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cestele S, Qu Y, Rogers J C, Rochat H, Scheuer T, Catterall W A. Neuron. 1998;21:919–931. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 36.Bezanilla F. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- 37.Wagner S, Lerche H, Mitrovic N, Heine R, George A L, Lehmann-Horn F. Neurology. 1997;49:1081–1025. doi: 10.1212/wnl.49.4.1018. [DOI] [PubMed] [Google Scholar]
- 38.Mitrovic N, George A L, Jr, Rüdel R, Lehmann-Horn F, Lerche H. Brain. 1999;122:1085–1092. doi: 10.1093/brain/122.6.1085. [DOI] [PubMed] [Google Scholar]