

Abstract
Engraftment of allogeneic bone marrow (BM) has been shown to induce tolerance to organs genotypically matched with the BM donor. Immune reconstitution after BM transplantation therefore involves re-establishment of a T cell pool tolerant to antigens present on both donor and host tissues. However, how hematopoietic grafts exert their influence over the regenerating immune system is not completely understood. Prior studies suggest that education of the newly arising T cell pool involves distinct contributions from donor and host stromal elements. Specifically, negative selection is thought to be mediated primarily by donor BM-derived antigen-presenting cells, whereas positive selection is dictated by radio-resistant host-derived thymic stromal cells. In this report we studied the effect of highly purified allogeneic hematopoietic stem cells (HSCs) on organ transplantation tolerance induction and immune reconstitution. In contrast to engraftment of BM that results in near-complete donor T cell chimerism, HSC engraftment results in mixed T cell chimerism. Nonetheless we observed that HSC grafts induce tolerance to donor-matched neonatal heart grafts, and one way the HSC grafts alter host immune responses is via deletion of newly arising donor as well as radiation-resistant host T cells. Furthermore, using an in vivo assay of graft rejection to study positive selection we made the unexpected observation that T cells in chimeric mice rejected grafts only in the context of the donor MHC type. These latter findings conflict with the conventionally held view that radio-resistant host elements primarily dictate positive selection.
Keywords: bone marrow transplantation, MHC restriction, mice
Transplantation of allogeneic bone marrow (BM) is known to alter immune responses in recipients so that tolerance is established to tissues matched with the genotype of the BM donors (1–3). Thus, the process of regeneration of the hematopoietic system involves the re-establishment of parameters that identify self- from nonself-antigens. The way in which BM grafts affect these changes is not completely understood. However, because T cells control antigen-specific immune responses the pathways that lead to regeneration of the peripheral T cell pool are central to immune reconstitution. T cell development after BM transplantation (BMT) is thought to recapitulate normal T cell ontogeny, which begins with the migration of BM-derived hematopoietic stem cells (HSCs) or more differentiated progenitors to the thymus (4). Within the thymus, under the influence of a specialized stromal microenvironment, progenitor T cells expand, differentiate, and undergo the rigorous processes of positive and negative selection (5–8). Positive selection results in survival of T cells with antigen receptors that corecognize self-MHC molecules plus foreign peptides. T cells whose receptors do not detect self-MHC molecules die, presumably by failure to receive critical differentiating signals. Negative selection involves the removal of potentially autoreactive T cells that interact too well with self-MHC molecules plus self-peptides.
Classic BM and thymus grafting studies by Zinkernagel et al. (9) and Bevan and Fink (10, 11) showed that the radio-resistant elements in the host thymus dictate MHC restriction of killer T cells. They proposed, and many experiments followed to support, the notion that these positively selecting elements in the thymus are epithelial cells (5, 6, 8). Subsequent studies refined these observations by tracking T cell development via expression of Vβ type or expression of a single transgenic T cell receptor and showed that both CD8+ and CD4+ T cells are likely to be positively selected on a subpopulation of epithelial cells located in the thymic cortex (5, 6, 8). In contrast, negative selection primarily is mediated by BM-derived antigen-presenting cells (APCs) (7, 12, 13). The absoluteness with which these stromal components dictate the selection processes continues to be challenged by discordant observations (14–17). In the setting of an MHC-mismatched allogeneic BMT, this schema of T cell selection predicts that the resultant host will be immunodeficient, insofar as the developing cells will be educated in the thymus to respond to antigens in the context of host MHC type, but will encounter BM-derived APCs in the periphery with the donor MHC type.
In the studies presented here we examined the issues of tolerance induction and immune reconstitution after transplantation of highly purified MHC-disparate HSCs in mice. HSCs are devoid of contaminating differentiated cell populations and thus, unlike most radiation BM chimeras, the effects of the donated immune system that arises from the HSC grafts are solely the result of de novo hematopoiesis. The HSC-transplanted mice also differ from BM chimeras because the former retain a significant proportion of radio-resistant host T cells (18). We found that HSCs induce tolerance to donor-matched organs and that such grafts can mediate negative selection of both developing donor T cells and residual T cells from the host. Furthermore, we made the unexpected observation that analysis of MHC restriction by an in vivo assay suggests that in chimeric mice the donor, not the host-type MHC, predominates in controlling heart graft rejection, a measure of T cell responsiveness. These studies, and the studies by Zinkernagel and Althage (17), reopen the issues of how, where, and on which cell types developing T cells learn MHC restriction and suggest that immunoincompetence in the post-BMT setting, a known clinical problem, is not completely explained by disparity between the MHC type of the donor versus the host.
Materials and Methods
Mice.
Three different C57BL/Ka congenic mouse lines were used as donors or recipients. C57BL/Ka were mice H-2b, Thy-1.2, CD45.2; congenic Thy-1.1 mice were H-2b, Thy-1.1, CD45.2 (C57BL/Ka.Thy-1.1) and designated as BA throughout the text; and congenic CD45.1 mice were H-2b, Thy-1.1, CD45.1 (C57BL/Ka.Thy-1.1.CD45.1) and designated BA.CD45.1 throughout the text. HSC or BM recipients were 7- to 10-week-old BALB/c (H-2d, Thy 1.2), BALB/k (H-2k, Thy 1.2), or C57BL/Ka mice. HSC and BM donors were BA or AKR/J mice (H-2k, Thy 1.1). For the neonatal heart transplantation experiments donors were 1- to 24-h-old neonates derived from BA, BA.CD45.1, BALB/c, C3H.SW (H-2b) or DBA.2 (H-2d) strain mice. All mice were bred and maintained at Stanford University.
HSC Purification.
Purified HSCs were obtained by modification of the methods described by Spangrude et al. (19). BM cells were positively selected for Sca-1 by using the MiniMACS/MidiMACS separation system (Miltenyi Biotech, Auburn, CA). The protocol was modified in January 1999 by the use of c-Kit instead of Sca-1 as the positively selecting marker. The c-Kit-enriched fraction was stained with FITC-conjugated α-Thy1.1 (19xE5), Texas red-conjugated α-Sca-1 (E13–161), allophycocyanin-conjugated α-c-Kit (2B8), and a mixture of phycoerythrin-conjugated lineage-specific mAbs as follows: α-B220 (6B2), α-CD3 mAb (145–2C11), α-CD5 (53–7.8), α-CD4 (GK-1.5), α-CD8 (53–6.7), GR-1 (8C5), α-Mac-1 (M1/70), and α-TER119 (TER199). The mAbs used for cell labeling were grown from hybridomas obtained from the American Type Culture Collection and conjugated to biotin or fluorochromes in our laboratory. The exceptions were the α-CD3 mAb (145–2C11) and the α-CD8 mAb (53–6.7) obtained from PharMingen. Cells were suspended in 1 mg/ml propidium iodide (Sigma) to allow exclusion of dead cells. The labeled cells were analyzed and sorted on a dual laser FACS (Becton Dickinson Immunocytometry Systems) made available through the FACS shared-user group at Stanford University. After sorting for FITClo, Texas redhi, allophycocyaninhi, and phycoerythrin-/lo, the purity of the resultant Thy-1loLin-/loSca+c-Kit+ cells was checked by FACs reanalysis, and sorted cells were >99% pure.
HSCs and BMT.
Recipient mice were conditioned for transplantation using split-dose lethal irradiation. BALB/c mice received 800 cGy and C57BL/Ka mice received 920 cGy delivered in two fractions. Some mice were conditioned for transplantation by using antibodies directed against natural killer determinants (α-AsialoGM1; Wako Chemical, Dallas, TX) and/or mAbs directed against the CD40 ligand (α-GP39) (a gift from R. Noelle, Dartmouth Medical School, Hanover, NH). HSCs or BM were injected by tail vein into mice within 24 h after irradiation.
Analysis of Peripheral Blood for Chimerism and Detection of Vβ+ Subsets.
Hematolymphoid chimerism was assessed by two-color immunofluorescence of peripheral blood leukocytes at ≈6 weeks after HSC transplantation and subsequently every 2–4 months to assess graft stability. Donor versus host cells were differentiated by mAbs specific for the MHC class I of donor mice (H-2b or H-2k) and double-stained with lineage-specific markers for B cells (α-B220, RA3–6B2), T cells (α-Thy-1.1 versus α-Thy-1.2, clones Ox-7 and 53–2.1, respectively), monocytes (α-Mac-1, M1/70.15), or granulocytes (α-GR.1, 8C5). Flow cytometry analysis was performed as described (18).
In selected mice the peripheral blood leukocytes were analyzed by FACS for the Vβ3+ (KJ25), Vβ6+ (RR4–7), or Vβ8+ (KJ16) subsets using biotin-conjugated α-Vβ mAbs and second-step labeling with streptavidin-FITC. The samples were double-stained with phycoerythrin-conjugated α-CD4 (H129–19) or phycoerythrin-conjugated α-CD3 (145–2C11). All mAb reagents were obtained from PharMingen except α-Mac-1, α-H-2b, and α-Vβ8+, which were obtained from Caltag (Burlingame, CA).
Intrapinna Neonatal Cardiac Transplants.
Newborn heart grafts were transplanted into the pinna of the ear of adult mice as described by Judd and Trentin (20). Donors were newborn mice 1–24 h old. Donor hearts from newborn mice were placed into the ear, and graft viability was assessed visually and by ECG activity.
Histologic Evaluation of Tissue from Chimeric Mice.
Selected chimeric mice were killed 4 months posttransplantation, and their thymuses and ears bearing neonatal heart grafts were snap-frozen in OCT compound (Tissue-Tek, Torrance, CA). Four-micrometer tissue sections were fixed in cold acetone and blocked as described (Vector Laboratories). Sections were stained with biotinylated antibodies to IAd, IAb, Thy 1.1, Thy 1.2, and Mac-1 (PharMingen), incubated with secondary antibody (streptavidin-horseradish peroxidase), visualized with 3-amino-9-ethylcarbazole as the chromagen, and counterstained with hematoxylin.
Mixed Lymphocyte Cultures.
Draining cervical lymph node responder cells were obtained from control or chimeric mice transplanted with neonatal heart grafts in the pinna of the ear and placed in single-cell suspension in RPMI supplemented with 10% FCS, l-glutamine, and β-mercaptoethanol. Stimulator cells were irradiated (3,300 rads) splenocytes from mice of the following strains: BA, BALB/c, C3H.SW, DBA.2, or BA into BALB/c chimeras. Triplicate samples of the different responder and stimulator cell combinations were plated at a concentration of 4 × 105 cells/well into 96-well flat bottom plates (Falcon, Becton Dickinson Labware) and cultured at 37°C with 5% CO2. The proliferative responses were evaluated by [3H]thymidine incorporation after 96 h.
Antibody Response to Sheep Red Blood Cells (SRBCs).
Hemolysin assays against SRBCs were performed as described (21). Mice were injected i.v. with 200 μl of a solution of 2.5% SRBCs in PBS, and serum was obtained by tail vein bleeding on day 9 postimmunization. Hemolysin titering was performed in 96-well U-bottom plates (Falcon, Becton Dickinson Labware). Each well received 25 μl of PBS, 25 μl of 1% SRBC solution, and 25 μl of titered serum, followed by a 30-min incubation at room temperature. A 1/16 dilution of guinea pig complement adsorbed with SRBCs was added followed by a 30-min incubation at 37°C. Titers were read as the last tube with evidence of hemolysis as shown by decreased “button” size and increased red color of clear fluid.
Results
Purified HSC Grafts Induce Tolerance to Donor-Matched Heart Grafts.
We previously established protocols to achieve long-term engraftment of purified HSCs transplanted across MHC barriers in mice (18). In the current studies chimeric BALB/c mice (H-2d) that were engrafted >2 months previously with MHC-disparate (BA) HSCs were transplanted with neonatal heart grafts genotypically matched with donor and host strain mice. Both donor and host-type grafts survived indefinitely (>100 days) (Table 1 and concurrent experiments reported elsewhere) (22). As controls, chimeric mice were challenged with third-party MHC-disparate grafts from C3H (H-2k) neonatal donors. Survival of third-party grafts was only slightly prolonged in chimeric mice as compared with control BALB/c mice, and all were rejected within 20 days. Other controls included BALB/c mice that received heart grafts from BALB/c or BA neonates. Grafts were rejected within 11–15 days (Table 1). In addition, chimeric BALB/c mice engrafted with BM from BA donors were similarly tolerant to donor and host strain neonatal heart grafts and rejected third-party C3H strain grafts although one of four did not reject the C3H graft. These data demonstrate that purified allogeneic HSC grafts can induce long-term tolerance to subsequently transplanted donor-matched heart grafts, and HSC recipients respond to third-party MHC-disparate grafts.
Table 1.
MHC-mismatched neonatal heart graft survival in chimeric mice
| Recipient | BALB/c graft survival, days | BA graft survival, days | C3H graft survival, days |
|---|---|---|---|
| Control BALB/c | 100 × 18 | 11 × 3, 13, 13 | 11 × 18 |
| BA HSC into BALB/c | >66*, >100 × 5 | 66†, >66*, >100 × 13 | 12 × 4, 13, 13, 15, 15 |
| BA BM into BALB/c | >100 × 5 | >100 × 7 | 11, 13, 13, >50 |
Data given as days × number.
Animal died from anesthesia.
Graft infected.
Chimerism Analysis of HSC-Engrafted Mice.
Donor hematopoietic cell chimerism in HSC- and BM-transplanted mice was assessed at ≈6 week postengraftment by FACS analysis of peripheral blood leukocytes. We observed that the HSC-transplanted mice were near-complete chimeras (>95%) in all white blood cell lineages except for the T lymphocytes. Table 2 shows the percent donor chimerism for T and B cells only. The results obtained for the macrophage and granulocytes lineages (data not shown) were similar to the B cell chimerism data. In HSC-transplanted mice, residual host Thy-1.2+ cells remained in the peripheral blood and comprised ≈30–40% of the total Thy-1+ population. The presence of significant numbers of residual host T cells is in contrast to mice transplanted with unmanipulated allogeneic BM that had significantly reduced residual host T cells. As we previously described, the likely explanation for this observation is that a nonstem cell population, presumably CD8+ cells, mediate an immune response that results in elimination of most residual host T cells (18, 23). Taken together with the neonatal heart graft data, the HSC chimeric mice were tolerant to donor alloantigens although they retained a significant proportion of host-derived T cells.
Table 2.
Percent chimerism of T and B cell subsets
| Experimental group | Thy-1.1+ (%), total Thy-1+ | H-2bB220+ (%), total B220+ |
|---|---|---|
| Control BA | 100 (n = 1) | 100 (n = 1) |
| Control BALB/c | 0 (n = 1) | 0 (n = 1) |
| BA HSC into BALB/c | 63 ± 3.8 (n = 3) | 100 (n = 3) |
| BA BM into BALB/c | 95 ± 4.0 (n = 3) | 97 ± 3.8 (n = 3) |
Values are expressed as mean ± SD.
Purified HSC Grafts Mediate Negative Selection of Donor and Host T Cells.
The effect of purified HSC grafts on T cell negative selection was studied by using mouse strain combinations with informative differences in endogenous superantigen expression. These differences allowed us to monitor deletion of donor- versus host-derived T cells. Donor strain AKR (H-2k) mice have integrated in their genome mouse mammary tumor virus proviruses including Mtv-7, which induces deletion of the Vβ6 subset, and others (24, 25). AKR mice therefore lack Vβ6+ T cells in their peripheral lymphoid organs and blood. Recipient strain mice with C57BL/Ka background genes have not integrated the Mtv-7 provirus and have measurable circulating levels of Vβ6+ cells (≈9% of CD4+ cells). In these studies the peripheral blood of chimeric mice was evaluated by FACS analysis 4 months posttransplantation. Donor and host T cells were distinguished by staining for H-2kCD3 and H-2bCD3. In BA mice engrafted with AKR HSC, donor AKR cells comprised ≈66% (65.6 ± 2.6) whereas residual host cells comprised ≈34% (34.4 ± 2.5) of CD3+ cells. Table 3 demonstrates that HSC grafts can mediate deletion of both donor and host T cells, because the Vβ6+ subset was virtually absent (<0.3% of CD4+ cells) from the blood of chimeric mice. Of note, host Vβ6+ T cells were presumably a radiation-resistant postthymic population, and thus the results suggest that HSC grafts mediate deletion of both newly arising and mature peripheral T cells. Vβ8 staining was used as a positive control and was present in chimeric mice at levels comparable to control mice. Reciprocal HSC transplantations using BA as donors and AKR mice as recipients demonstrated that radio-resistant host elements also mediate negative selection (Table 3).
Table 3.
Vβ deletion of donor- and host-derived peripheral T cells
| Experimental group | % Vβ6+ CD3+ | % Vβ6+ CD4+ | % Vβ8+ CD3+ | % Vβ8+ CD4+ |
|---|---|---|---|---|
| Control BA | 8.99 ± 0.75 (n = 4) | 9.17 ± 0.09 (n = 3) | 17.18 ± 0.17 (n = 4) | 18.73 ± 0.27 (n = 3) |
| Control AKR | 0.94 ± 1.1 (n = 2) | 0.08 ± 0.02 (n = 2) | 11.70 ± 0.85 (n = 2) | 11.51 (n = 1) |
| BA HSC into AKR | 1.67 ± 0.12 (n = 2) | ND | 9.20 ± 3.68 (n = 2) | ND |
| AKR HSC into BA | 0.36 ± 0.07 (n = 4) | 0.24 ± 0.1 (n = 4) | 16.53 ± 1.26 (n = 4) | 17.49 ± 0.68 (n = 4) |
ND, not determined.
To confirm these findings we studied T cell deletion in a strain combination where both donor and host are known to delete different Vβ subsets. Donors were AKR (H-2k) mice and recipients were BALB/k (H-2k), a genetic mismatch comparable to matched unrelated donor transplants in human. Deletion of both Vβ6+ and Vβ3+ T cells depends on MHC class II I-E expression, and both strains in this experiment express I-E. T cells between the two strains were delineated by Thy-1 allelic markers because AKR mice were Thy-1.1+ and BALB/k mice were Thy-1.2+. BALB/k mice delete the Vβ3 subsets, which comprise <0.3% of their peripheral CD4+ cells, but they have circulating Vβ6+ cells at a level of ≈10% of their peripheral CD4+ cells. In contrast, AKR mice have ≈8% Vβ3+CD4+ peripheral T cells, and as noted above, they delete the Vβ6 subset. Peripheral blood analysis performed on chimeric mice at ≈8 months posttransplantation revealed that ≈15% (14.7 ± 6.4) of CD3+ T cells in HSC chimeric mice were of BALB/k (host) origin. As shown in Table 4, transplantation of purified AKR HSCs into BALB/k mice resulted in chimeric mice that lacked both Vβ3+ and Vβ6+ T cells.
Table 4.
Reciprocal deletion of donor and host Vβ T cell subsets derived peripheral T cells
| Experimental group | % Vβ3+ CD4+ | % Vβ6+ CD4+ |
|---|---|---|
| Control AKR | 8.0 (n = 1) | 0.06 (n = 1) |
| Control BALB/k | 0.02 (n = 1) | 10.55 (n = 1) |
| AKR HSC into BALB/k | 0.11 ± 0.06 (n = 10) | 0.07 ± 0.02 (n = 10) |
Values are expressed as mean ± SD.
Thymuses from HSC Chimeric Mice Contain High Levels of Donor MHC Class II+ Cells.
The functional studies described above indicated that elements derived from purified HSC grafts mediate negative selection. It has been previously reported that hematolymphoid-derived thymic APCs play a central (7, 12, 13), although not exclusive (26–28), role in negative selection of developing T cells. Therefore, we examined the thymuses of HSC chimeras 4–6 months posttransplantation for evidence of APCs derived from HSC donors (I-Ab) versus hosts (I-Ad) using immunohistochemistry staining for class II MHC molecules. Fig. 1B shows an abundance of interdigitating cells located throughout the thymic cortex and medulla of an HSC chimeric mouse that stain positively for donor I-Ab. Host type (I-Ad) dendritic cells also were found at low levels in the thymuses of the chimeras (Fig. 1B); the epithelial versus hematolymphoid origin of these cells is unknown. Staining for donor (Thy-1.1) and host-derived (Thy-1.2) thymocytes also showed a predominance of donor-type cells, although residual host thymocytes were seen to persist even 4–6 months after lethal irradiation and HSC transplantation (Fig. 1 C and D).
Figure 1.
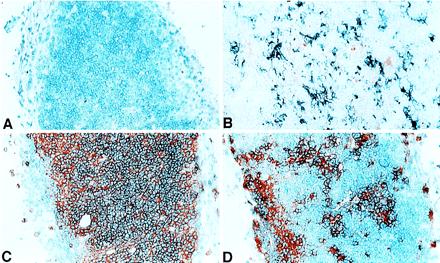
Immunohistochemical stains of donor versus host T cells and MHC class II+ cells (APCs) in the thymus of allogeneic HSC chimeric mice. (A) Background staining with control second-step reagent. (B) Double staining for donor BA MHC class II+ cells (IAb in blue) versus host BALB/c MHC class II+ cells (IAd, in red). (C) Single stain in red for donor T cells (Thy-1.1). (D) Single stain in red for recipient T cells (Thy-1.2).
Progeny of Allogeneic HSCs Mediate Positive Selection.
To determine the functionally predominant MHC type after HSC transplantation, long-term chimeras (BA into BALB/c) were challenged with neonatal heart grafts that were matched at the MHC loci of either the donor or host strain but that differed in background minor histocompatibility (mH) genes. Graft function was assessed by visual inspection and ECG analysis. DBA.2 mice are H-2d at the MHC and thus matched with the recipient strain BALB/c mice and differ at mH loci. Likewise, C3H.SW mice are H-2b at the MHC and matched with the donor strain BA mice but differ at mH loci. T cells that reject DBA/2 (H-2d) grafts presumably would have been positively selected by the H-2d thymic elements, whereas cells that reject CSH.SW (H-2b) grafts presumably would have been positively selected by H-2b thymic elements. Control unmanipulated mice that received mH-mismatched grafts reject these grafts in ≈2 weeks (Table 5). In contrast, the BA into BALB/c HSC chimeras slowly rejected C3H.SW (H-2b) grafts and did not reject DBA.2 (H-2d) grafts. Rejection was documented in six of eight C3H.SW (H-2b) grafts and none of eight DBA.2 (H-2d) grafts. Furthermore, the times to rejection of C3H.SW grafts were greatly prolonged because most of these grafts were rejected between 50 and 100 days posttransplantation (Table 5). Selected mice were killed 7–12 weeks posttransplantation for histologic analysis of neonatal heart grafts. The morphologic evidence agreed with the functional data and showed intact myocardium with minimal mononuclear cell infiltrates in the DBA.2 grafts, whereas the CSH.SW grafts had decreased amounts of myocardial tissue with high levels of mononuclear and T cell infiltration (Fig. 2).
Table 5.
MHC-mismatched neonatal heart graft survival in chimeric mice
| Recipient | BALB/c graft survival, days | DBA/2J graft survival, days | C3H.SW graft survival, days |
|---|---|---|---|
| Control BALB/c | >120 × 18 | 11 × 4, 12, 13 × 5, 23 | 11 × 5, 12 × 3, 13 × 2 |
| Control BA | ND | 11, 12 × 3, 14 × 5 | 9, 11 × 4, 13 × 2, 15 |
| BA HSC into BA.CD45.1 | ND | ND | 11 × 3, 15 × 3 |
| BA HSC into BALB/c | >66*, >120 × 5 | >82† × 2, >104, >118, >119*, >125, >139 × 2 | 51† × 2, 62 × 2, >84*, 97, >99, 117 |
| BA BM into BALB/c | >120 × 5 | >61*, >92†, >103, 119, >139 | >48*, 65, 70, >79†, 86 |
Data given as days × number. ND, not determined.
Animal died from anesthesia.
Grafts taken for immunohistochemistry.
Figure 2.
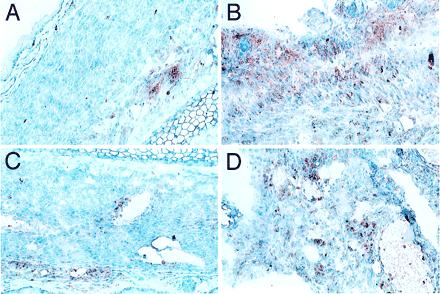
Immunohistochemical analysis of mH-mismatched neonatal heart grafts transplanted in HSC chimeric mice. A BALB/c mouse engrafted with BA HSCs was killed on day +82 and day +51 after transplantation of heart grafts from a DBA.2 (A and C) and a C3H.SW (B and D) neonate into the pinna of contralateral ears, respectively. (A and B) Staining for macrophages and monocytes (α-Mac-1). (C and D) Staining for T cells (α-CD3). Note healthy appearance of myocardial tissue and mild peripheral involvement with inflammatory cells in the DBA.2 graft as compared with the C3H.SW, which has heavy mononuclear cell involvement and evidence of dying myocardial cells.
It was possible that the impaired ability of chimeric mice to recognize and reject mH-mismatched grafts was caused by the HSC transplant procedure itself rather than unique aspects of immune reconstitution after an allogeneic hematopoietic cell transplantation. Therefore, BA mice that had been engrafted >6 weeks previously with BA.CD45.1 congenic HSCs were tested for their ability to reject mH-mismatched C3H.SW neonatal heart grafts. All six of these mice rejected the C3H.SW grafts in 11–15 days (Table 5) and permanently accepted CD45 congenic grafts (data not shown). Taken together these data show that mice engrafted with MHC-disparate HSCs or BM slowly reject third-party heart grafts that are MHC-matched with the hematopoietic cell donor but do not reject grafts MHC-matched with the host. The results suggest that minor transplantation antigens are seen in the context of donor MHC type and thus MHC restriction, the outcome of thymic positive selection, is dictated in these chimeras by the hematopoietic graft.
In Vitro T Cell Proliferative Responses Are H-2d Restricted, Whereas Graft Rejection Is H-2b Restricted.
In vitro mixed lymphocyte reactions were performed to confirm that positive selection occurs on the MHC type of the stem cell donor rather than the host. Cells from the draining cervical lymph nodes from BA into BALB/c chimeric mice that had been sensitized in vivo with C3H.SW and DBA.2 heart grafts were used as responders. Stimulator cells were BA, BALB/c, C3H.SW, or DBA.2 splenocytes. As shown in Fig. 3, all lymph node cells from chimeric mice made proliferative responses against DBA.2 splenocytes, but not against C3H.SW, BA, or BALB/c splenocytes. It was also of interest that BM chimeras as compared with HSC chimeras had significantly reduced responses against DBA.2 splenocytes (P < 0.02). Thus, in contrast to the in vivo data, which suggest that the predominant restricting MHC type in the chimeric mice is donor-derived (H-2b), the in vitro data show the opposite and support the notion that the predominant restricting MHC element is derived from the host (H-2d). Thus, we conclude that assessment of T cell function by mixed lymphocyte reactions may not serve as an accurate surrogate assay for systemic T cell immunity as measured by heart graft rejection.
Figure 3.
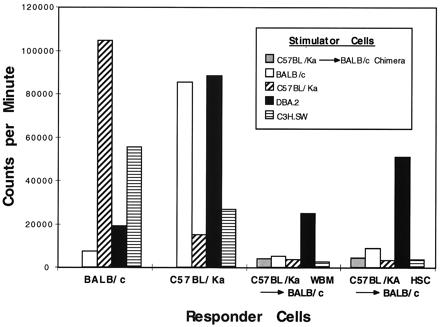
Mixed lymphocyte responses of allogeneic HSC or BM chimeras against third-party mH mismatched stimulators. Lymph node cells from control BALB/c and BA mice make robust responses to MHC-disparate splenocytes and reduced, but measurable, responses to mH-disparate splenocytes (DBA.2 and C3H.SW, respectively). In contrast, hematopoietic cell chimeras respond only against the DBA.2 splenocytes.
Allogeneic HSC Chimeras Can Mount Primary Antibody Responses to SRBCs.
The BA into BALB/c HSC or BM chimeras efficiently rejected third-party MHC-disparate hearts but showed prolonged times to rejection of C3H.SW hearts and could not reject DBA.2 hearts. To study whether the blunted response to mH-disparate grafts revealed deficiencies in helper T cell generation and function, the T cell-dependent ability to produce antibodies to SRBCs was tested. Chimeric mice were immunized with SRBCs at >3 months posttransplantation. As shown in Table 6 the day 9 SRBC response in allogeneic HSC-transplanted mice was equivalent to unmanipulated control BALB/c mice. In addition, mice transplanted with allogeneic BM had evidence of reduced titers to SRBCs as compared with HSC-transplanted (P < 0.07) and control mice (P < 0.05).
Table 6.
Anti-SRBC response of chimeric mice
| Experimental group | Preimmune SRBC titer, mean log2 ± SD | Day 9 SRBC titer, mean log2 ± SD |
|---|---|---|
| Control BALB/c | 0 ± 0 (n = 4) | 7.5 ± 0.58 (n = 4) |
| BA HSC into BALB/c | 0 ± 0 (n = 4) | 7.3 ± 0.50 (n = 4) |
| BA BM into BALB/c | 0 ± 0 (n = 4) | 5.5 ± 1.29 (n = 4) |
Discussion
In this report we studied whether purified allogeneic HSCs will induce tolerance to donor-matched solid organs and examined the way in which these grafts alter recipient immune responses. We determined that engraftment of MHC-mismatched HSCs did indeed induce tolerance to heart grafts when the graft was matched with the HSC donor, and that HSC chimeric mice were able to reject third-party MHC-mismatched grafts. The stem cell graft mediated negative selection of potentially donor reactive T cells. In fact, both donor and radio-resistant host elements mediated negative selection, because peripheral T cells in the chimeras were devoid of the Vβ subsets deleted in both donor and host strain mice.
In our studies wherein chimeric mice were challenged with heart grafts matched at the MHC of the donor versus the host, but differing with regard to mH antigens, we made the unexpected observation that only the grafts of the donor-type MHC were rejected, albeit slowly. This test of systemic T cell immunity can be interpreted as demonstrating that post-BMT the functional pool of T cells recognize minor antigens in the context of the MHC of the hematopoietic cell donor but not the host. These results conflict with the widely held view that positive selection of T cells occurs exclusively on the radio-resistant thymic epithelial cells of the host (5, 8–11). Recent studies by Zinkernagel et al. (17) and Zerrahn et al. (16) also have questioned the relative importance of the thymic epithelium in dictating positive selection, as compared with hematopoietic-derived cells. They demonstrated that responses measured by in vitro analysis can be restricted to the BM donor MHC type when BM from normal or T cell antigen receptor transgenic mice was transplanted into thymectomized nude or class I-deficient mice (16, 17). Our studies show that in an in vivo assay of graft rejection the predominant MHC restricting element is from the donor hematopoietic cells.
In earlier studies (29, 30) CD4 T cell positive selection was proposed to be restricted mainly by radio-resistant thymic epithelial cells, whereas positive selection of CD8+ T cells could be mediated in part by hematopoietic-derived cells. Using MHC class II-deficient mice to generate chimeras with tissue-selective expression of MHC class II, Markowitz et al. (30) found that CD4+ cells developed only when class II MHC molecules were expressed on radio-resistant thymic cells. In contrast, Bix and Raulet (14) showed that positive selection of CD8+ T cells can occur on class I-expressing hematopoietic cells, although the efficiency appeared to be substantially lower than with class I-positive resident thymic cells. Our own data demonstrate an interesting dichotomy. In vitro analysis of HSC chimera lymph node cells by mixed lymphocyte reactions—an assay that is mainly CD4 T cell-driven—showed proliferative responses only against minor antigens presented by the host type MHC; whereas as noted above, organ rejection occurred only if the minor antigens were presented by the HSC donor type MHC. Because the neonatal heart grafts were replete with resident APCs these differences are not explainable by the lack of APCs in the in vivo versus the in vitro assay. One explanation for the in vitro T cell proliferative response against DBA.2 splenocytes is that this strain expresses the Mtv-7 superantigen, whereas none of the other strains used as stimulators express this antigen and therefore have not deleted Mtv-7 in vitro reactive cells. Although it is also conceivable that residual BALB/c radio-resistant T cells directly recognize and are responsible for the rejection of H-2b C3H.SW heart grafts, such cells would have to recognize minor C3H.SW antigens as well, because the vast majority of blood cells are H-2b (BA).
The HSC chimeric mice can reject third-party MHC-disparate heart grafts and mount antibody responses against SRBCs. However, the delay or absence of responses against mH antigens demonstrates the degree to which these mice are immunodeficient. It has been argued that such mice will be immunodeficient because of the genotypic disparity between the donor and host cell populations responsible for negative and positive selection, respectively. However, as discussed above, we and others have shown that hematopoietic-derived elements direct both positive and negative selection and in some circumstances may be the primary mediators of T cell selection. Zinkernagel and Althage (17) have proposed that the thymic environment provides a milieu that supports T cell differentiation and receptor expression of young thymocytes, whereas hematopoietic cells dictate T cell specificity. Thus, a central issue requiring further examination is to determine whether the long-term immunodeficiency after allogeneic hematopoietic cell transplantation can be entirely explained by inefficiencies in positive selection or if other factors lead to impairment of T cell responses. These other factors include T cell education in a small and compromised thymus, blunted T cell responses caused by extra-thymic development, or a T cell regenerative process that favors negative selection. Here we also observed that chimeras receiving allogeneic BM versus HSCs were relatively more immunodeficient as judged by the mixed lymphocyte reactions assay and the response to SRBCs. Because the vast majority of cells in BM are not HSCs, it is possible that populations contained in the BM inoculum exerts an additional immunosuppressive effect (e.g., subclinical graft-vs.-host disease) (31). The experience with human allogeneic BMT argues that MHC mismatch alone is not the only cause for long-term immunodeficiency in the posttransplantation setting. The vast majority of BMT patients receive grafts from HLA identical donors, yet are highly susceptible to infectious complications even after the graft is established and in the absence of confounding factors such as pharmacologic immunosuppression or clinically evident graft-vs.-host disease (32, 33).
Finally, our studies directly address issues in clinical solid organ transplantation. Donor organs are a limited resource and thus for patients receiving cadaver grafts there is little or no matching of HLA antigens. The approach of using BM to induce tolerance to donated organs has been a theoretical possibility for decades; however, its application to clinical practice has been limited by the risks of the allogeneic BMT procedure (33). The largest risk has been graft-vs.-host disease, which is caused by T cells that accompany an unmanipulated BM graft. The technologies of cell purification (34–36) now have provided ways to transplant highly purified stem cell grafts devoid of the graft-vs.-host disease-inducing cell populations. We showed here that purified HSCs tolerize the recipient to donor-matched organs. We anticipate that in the future trials of organ tolerance induction using purified hematopoietic grafts will be performed. In our view, the remaining obstacle will be understanding and perhaps manipulating immune reconstitution in the posttransplant setting. Based on our data we believe that it is critical to study this issue in animals and follow their responses to pathogens with in vivo readouts to accurately predict the effect that manipulations of the hematopoietic system will have on transplant patients.
Acknowledgments
We thank Mr. Tim Knaak for his assistance with cell sorting, Ms. Randy Shorthouse for her assistance with the neonatal heart transplantations, Mr. Phil Verzola for production of the figures, and Ms. Sarah Clark for her secretarial assistance. This work was supported by the Burroughs Wellcome Fund (J.A.S.), a National Cancer Institute award (I.L.W., #R535CA2551), and National Institutes of Health Grants PO1-DK53005–01 and 2PO1-CA49605, and aided by institutional support by the Howard Hughes Medical Institute (J.A.S.).
Abbreviations
- HSC
hematopoietic stem cell
- mH
minor histocompatibility
- BM
bone marrow
- BMT
BM transplantation
- APC
antigen-presenting cell
- SRBC
sheep red blood cell
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170279297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170279297
References
- 1.Billingham R E, Brent L, Medawar P B. Nature (London) 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 2.Charlton B, Auchincloss H, Jr, Fathman C G. Annu Rev Immunol. 1994;12:707–734. doi: 10.1146/annurev.iy.12.040194.003423. [DOI] [PubMed] [Google Scholar]
- 3.Wekerle T, Sykes M. Transplantation. 1999;68:459–467. doi: 10.1097/00007890-199908270-00001. [DOI] [PubMed] [Google Scholar]
- 4.Aguila H L, Akashi K, Domen J, Gandy K L, Lagasse E, Mebius R E, Morrison S J, Shizuru J, Strober S, Uchida N, et al. Immunol Rev. 1997;157:13–40. doi: 10.1111/j.1600-065x.1997.tb00971.x. [DOI] [PubMed] [Google Scholar]
- 5.Benoist C, Mathis D. In: Fundamental Immunology. Paul W, editor. Philadelphia: Lippincott–Raven; 1999. pp. 367–409. [Google Scholar]
- 6.von Boehmer H. Cell. 1994;76:219–228. doi: 10.1016/0092-8674(94)90330-1. [DOI] [PubMed] [Google Scholar]
- 7.Sprent J, Webb S R. Curr Opin Immunol. 1995;7:196–205. doi: 10.1016/0952-7915(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 8.Moller G. Immunol Rev. 1993;135:5–242. [Google Scholar]
- 9.Zinkernagel R M, Callahan G N, Klein J, Dennert G. Nature (London) 1978;271:251–253. doi: 10.1038/271251a0. [DOI] [PubMed] [Google Scholar]
- 10.Fink P J, Bevan M J. J Exp Med. 1978;148:766–775. doi: 10.1084/jem.148.3.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevan M. Nature (London) 1977;269:417–418. [Google Scholar]
- 12.Matzinger P, Guerder S. Nature (London) 1989;338:74–76. doi: 10.1038/338074a0. [DOI] [PubMed] [Google Scholar]
- 13.Blackman M, Kappler J, Marrack P. Science. 1990;248:1335–1341. doi: 10.1126/science.1972592. [DOI] [PubMed] [Google Scholar]
- 14.Bix M, Raulet D. Nature (London) 1992;359:330–333. doi: 10.1038/359330a0. [DOI] [PubMed] [Google Scholar]
- 15.Longo D L, Schwartz R H. Nature (London) 1980;287:44–46. doi: 10.1038/287044a0. [DOI] [PubMed] [Google Scholar]
- 16.Zerrahn J, Volkmann A, Coles M C, Held W, Lemonnier F A, Raulet D H. Proc Natl Acad Sci USA. 1999;96:11470–11475. doi: 10.1073/pnas.96.20.11470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zinkernagel R M, Althage A. Proc Natl Acad Sci USA. 1999;96:8092–8097. doi: 10.1073/pnas.96.14.8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shizuru J A, Jerabek L, Edwards C T, Weissman I L. Biol Blood Marrow Transplant. 1996;2:3–14. [PubMed] [Google Scholar]
- 19.Spangrude G J, Heimfeld S, Weissman I L. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 20.Judd K P, Trentin J J. Transplantation. 1971;11:298–303. [PubMed] [Google Scholar]
- 21.Eisenberg R A, Weissman I L. J Immunol. 1971;106:143–149. [PubMed] [Google Scholar]
- 22.Gandy K L, Weissman I L. Transplantation. 1998;65:295–304. doi: 10.1097/00007890-199802150-00001. [DOI] [PubMed] [Google Scholar]
- 23.Gandy K L, Domen J, Aguila H, Weissman I L. Immunity. 1999;11:579–590. doi: 10.1016/s1074-7613(00)80133-8. [DOI] [PubMed] [Google Scholar]
- 24.Marrack P, Winslow G M, Choi Y, Scherer M, Pullen A, White J, Kappler J W. Immunol Rev. 1993;131:79–92. doi: 10.1111/j.1600-065x.1993.tb01531.x. [DOI] [PubMed] [Google Scholar]
- 25.Acha-Orbea H, Held W, Waanders G A, Shakhov A N, Scarpellino L, Lees K R, MacDonald H R. Immunol Rev. 1993;131:5–25. doi: 10.1111/j.1600-065x.1993.tb01527.x. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann M W, Heath W R, Ruschmeyer D, Miller J F A P. Proc Natl Acad Sci USA. 1995;92:9851–9855. doi: 10.1073/pnas.92.21.9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Speiser D E, Pircher H, Ohashi P S, Kyburz D, Hengartner H, Zinkernagel R M. J Exp Med. 1992;175:1277–1283. doi: 10.1084/jem.175.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oukka M, Colucci-Guyon E, Tran P L, Cohen-Tannoudji M, Babinet C, Lotteau V, Kosmatopoulos K. Immunity. 1996;4:545–553. doi: 10.1016/s1074-7613(00)80481-1. [DOI] [PubMed] [Google Scholar]
- 29.Bradley S M, Kruisbeek A M, Singer A. J Exp Med. 1982;156:1650–1664. doi: 10.1084/jem.156.6.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markowitz J S, Auchincloss H, Jr, Grusby M J, Glimcher L H. Proc Natl Acad Sci USA. 1993;90:2779–2783. doi: 10.1073/pnas.90.7.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shearer G. Immunol Rev. 1983;73:115–126. doi: 10.1111/j.1600-065x.1983.tb01081.x. [DOI] [PubMed] [Google Scholar]
- 32.Small T, Papdopoulous E, Boulad F, Black P, Castro-Malaspina H, Childs B, Collins N, Gillio A, George D, Jakubowski A, et al. Blood. 1999;93:467–480. [PubMed] [Google Scholar]
- 33.Thomas E D, Blume K G, Forman S J, editors. Hematopoietic Cell Transplantation. Malden, MA: Blackwell Sciences; 1999. [Google Scholar]
- 34.Baum C M, Weissman I L, Tsukamoto A S, Buckle A M, Peault B. Proc Natl Acad Sci USA. 1992;89:2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Archimbaud E, Philip I, Coiffier P, Michallet M, Salles G, Sebban C, Roubi N, Lopez F, Bessueille L, Mazars P, et al. Blood. 1997;90:394a. [Google Scholar]
- 36.Negrin R, Atkinson K, Leemhuis T, Hanania E, Juttner C, Tierney K, Hu W, Johnston L, Shizuru J, Stockerl-Goldstein K, et al. Biol Blood Marrow Transplant. 2000;6:262–271. doi: 10.1016/s1083-8791(00)70008-5. [DOI] [PubMed] [Google Scholar]