

Abstract
We report the identification of a TRAF-interacting protein, T6BP, that specifically associates with TRAF6. This interaction occurs between the coiled-coil region of T6BP and the N-terminal ring finger and zinc finger domains of TRAF6. IL-1, but not tumor necrosis factor, induces TRAF6–T6BP complex formation in a ligand-dependent manner. Formation of the TRAF6–T6BP complex depends on the presence of the IL-1 receptor-associated kinase (IRAK). After IL-1 stimulation, TRAF6 can exist in two separate complexes, TRAF6–IRAK or TRAF6–T6BP, but IRAK is not present in TRAF6–T6BP complexes. T6BP does not seem to play a direct role in activation of IκB kinases or Jun N-terminal kinase.
Tumor necrosis factor receptor (TNFR)-associated factors (TRAFs) are a family of proteins that were discovered initially as downstream signal transducers of the TNFR superfamily (1–3). More recently, TRAFs were also shown to be involved in signaling by Toll/IL-1 receptor (IL-1R) family members (4, 5). To date, six members of the TRAF family have been identified (1–4, 6–11). TRAFs 1–6 are characterized by a coiled-coil TRAF-N domain and a conserved C-terminal TRAF-C domain (4). The TRAF-C domain mediates interactions among TRAF proteins as well as their association with members of the TNFR superfamily (3, 12, 13). In addition, TRAFs 2–6 contain an N-terminal ring finger/zinc finger region that is thought to be essential for downstream signaling (1, 3, 4, 9–11).
Several members of the TRAF family, including TRAF2, TRAF5, and TRAF6, have been implicated in various signal transduction pathways leading to the activation of the transcription factor NF-κB (4, 9–11, 14). Overexpression of TRAF2, TRAF5, or TRAF6 activates NF-κB, and truncated versions of TRAF2, TRAF5, and TRAF6 lacking zinc-binding domains act as dominant-negative inhibitors of NF-κB activation mediated by various stimuli, suggesting that these TRAFs are common mediators for NF-κB activation (4, 9–11, 14). On the other hand, TRAF1, TRAF3, and TRAF4 do not seem to be involved in NF-κB activation (15).
Data on the physiologic roles of four TRAF family members have been determined by gene targeting experiments (5, 16–20). TRAF2, TRAF3, and TRAF6 are essential for perinatal and postnatal survival (5, 16, 18). Furthermore, studies on TRAF6-deficient mice revealed that TRAF6 is required for IL-1-, CD40-, lipopolysaccharide-, and IL-17-induced NF-κB activation (5, 20). Moreover, TRAF6-deficient mice are osteopetrotic with defects in bone remodeling and tooth eruption caused by impaired osteoclast function (5).
Among the six TRAFs described to date, TRAF6 is the least homologous across the prototypical TRAF domain (4). TRAF6 was identified originally in 1996 as a molecule that binds to the cytoplasmic domain of CD40 (11) and that acts as a signal transducer in the IL-1 signaling pathway (4). The important role of TRAF6 in IL-1 signaling is now well established. IL-1 induces the complex formation of the type I receptor (IL-1RI) and the receptor accessory protein. The cytosolic protein MyD88 is then recruited to the receptor complex, where it acts as an adaptor protein and mediates the association of IL-1 receptor-associated kinase (IRAK) with the receptor (21–23). IRAK then interacts with TRAF6, which mediates IL-1-induced NF-κB and c-jun N-terminal kinase (JNK) activation (4, 5). The mechanism by which TRAF6 is then able to activate the kinases involved in the NF-κB and JNK pathways is not understood. The activation of NF-κB requires the IκB kinase complex (24), and the activation of JNK is thought to be mediated by the MEKK family of protein kinases (25).
To gain more insights about the physiologic roles of TRAF6, we performed a two-hybrid screening with a HeLa cell cDNA library and full-length TRAF6 as a bait. We identified a 86-kDa protein, T6BP, that associates with TRAF6 after IL-1 stimulation. T6BP seems to represent a previously unidentified component in IL-1 signaling.
Materials and Methods
Reagents.
Recombinant human IL-1β was purchased from BioSource International (Camarillo, CA). Recombinant human TNF was provided by Genentech. Anti-Flag monoclonal antibody M2 was purchased from Sigma. Rabbit anti-Flag, anti-Myc, anti-glutathione S-transferase (anti-GST), and anti-CD40 polyclonal antibodies were from Santa Cruz Biotechnology. Glutathione beads were purchased from Amersham Pharmacia. The anti-TRAF6 antibodies, anti-IRAK antibody, 293.IL-1RI cells, and I1A cells were kindly provided by Zhaodan Cao (Tularik). The 293.CD40 cells and a membrane preparation of CD40 ligand were generously provided by Ralf Schwandner (Tularik). The rabbit anti-T6BP antiserum was raised against a 34-mer peptide: ELKIEKTMKEKEELLKLIAVLEKETAQLREQVGR.
Expression Vectors.
Mammalian expression vectors encoding TRAFs 1–6 have been described (15). T6BP was expressed as N-terminally tagged Flag or Myc fusion proteins by using the pRK vector. Deletion mutants of T6BP and TRAF6 were generated by PCR.
Yeast Two-Hybrid Cloning.
DNA encoding full-length TRAF6 was used as bait in the MATCHMAKER LexA Two-hybrid System (CLONTECH). The two-hybrid screening was performed according to the manufacturer's recommendations, except that a pretransformed HeLa cDNA library (generously provided by Hsing-Jien Kung, University of California, Davis, CA) was used. Positive yeast clones were selected by prototrophy for leucine and expression of β-galactosidase. Yeast DNA was recovered and transformed into Escherichia coli. The cDNA insert from the two-hybrid clone of T6BP (which corresponds to the C-terminal part of the full-length T6BP) was used to search the expressed sequence tag database, and clone AI016700 encoding the full-length T6BP was purchased from Genome Systems (St. Louis). DNA sequencing was performed on an Applied Biosystems automated DNA sequencer.
Transfection, Immunoprecipitation, and Immunoblotting Analyses.
293 cells were transiently transfected with expression plasmids with the Profection calcium phosphate transfection kit (Promega). After 24–36 h, cells were washed with cold PBS and lysed in RIPA buffer containing 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4 (pH 7.2), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM NaF, and the Complete protease inhibitors (Roche Molecular Biochemicals). Immunoprecipitation and immunoblotting analyses were performed as described (26).
For detection of the endogenous interaction of TRAF6 with T6BP, 293.IL-1RI cells (a 293 cell line stably expressing IL-1RI; ref. 4), HeLa cells, 293.CD40 cells (a 293 cell line stably expressing Flag-tagged CD40), or I1A cells (IRAK-deficient 293.IL-1RI cells; ref. 27) were stimulated with 10 ng/ml IL-1, 100 ng/ml TNF, or 250 μg/ml membrane preparation of CD40 ligand at 37°C for various times. After extensive washing with cold PBS, cells were lysed in lysis buffer A (20 mM Tris·Cl, pH 7.5/150 mM NaCl/1% Triton X-100/1 mM EDTA/1 mM NaF/1 mM PMSF and the complete protease inhibitors; ref. 26). Immunoprecipitation was carried out with rabbit anti-T6BP antibody, and immunoblotting was performed with a monoclonal mouse anti-TRAF6 antibody or a polyclonal rabbit anti-T6BP antibody.
JNK Assays and Reporter Assays.
Kinase assays and reporter assays were performed as described (28). Briefly, for the JNK assay, 293 cells were transiently transfected with 1 μg of hemagglutinin (HA)-JNK plasmid (29) together with various amounts of TRAF6 or T6BP constructs. The total DNA transfected (10 μg) was kept constant by supplementation with control vector pRK5. Cell lysates were immunoprecipitated with anti-HA antibody (Roche Molecular Biochemicals) and subjected to kinase reaction with 1 μg of GST-c-Jun (amino acids 1–79) as substrate. For the reporter assays, 293 cells were transfected with an E-selectin-luciferase reporter gene plasmid, a β-galactosidase internal control plasmid, and various amounts of expression constructs. Reporter gene activity was determined with the Luciferase Assay System (Promega).
Results
Isolation of cDNA Clone Encoding T6BP.
We performed yeast two-hybrid interaction cloning to screen for proteins that associate directly with TRAF6. In this screening, we identified a TRAF6-binding protein, T6BP. The full-length T6BP has 747 amino acids and is almost identical to the previously described TXBP151-L (30), with a difference in 2 amino acids (amino acids 233 and 234 are glutamine and leucine, respectively, in T6BP and histidine and valine, respectively, in TXBP151-L; Fig. 1). TXBP151-L has been identified as a TAX- and A20-binding protein (30). T6BP has three coiled-coil domains (amino acids 185–248, 350–413, and 526–589) and two helix–loop–helix regions (amino acids 206–225 and 435–454) as identified by Genetics Computer Group protein analysis tools (Madison, WI; Fig. 1). T6BP shares an overall 50% identity and 59% similarity to a recently identified chicken protein MDP62, which has been implicated in promoting neurite outgrowth (31).
Figure 1.

Predicted amino acid sequence of T6BP. The amino acid sequence deduced from the nucleotide sequence of the full-length T6BP cDNA is shown. The single underlines show the coiled-coil regions. The 34-amino acid peptide used for generating anti-T6BP antibody is indicated by a double underline. The two amino acids that are different from TXBP151 are indicated by asterisks. The full-length T6BP nucleotide sequence has been deposited in GenBank (accession no. AF268075).
T6BP Specifically Interacts with TRAF6.
The interaction of T6BP with TRAF6 was analyzed in mammalian cell coimmunoprecipitation assays. Full-length T6BP containing an N-terminal Myc epitope tag was transiently coexpressed in 293 cells with Flag epitope-tagged TRAF proteins. Cell lysates were immunoprecipitated with an anti-Flag monoclonal antibody. Coprecipitating T6BP was detected by immunoblotting analysis with anti-Myc polyclonal antibodies. In this assay, T6BP specifically coprecipitates TRAF6 but not TRAFs 1–5 (Fig. 2).
Figure 2.
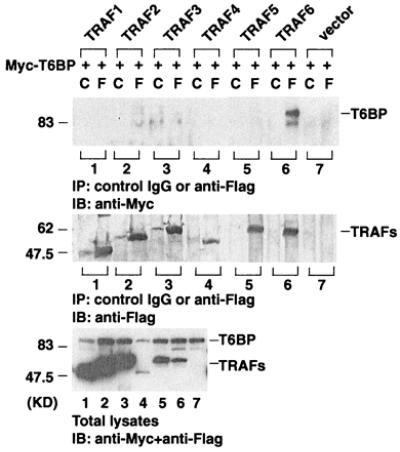
T6BP specifically interacts with TRAF6. 293 cells were transiently transfected with expression vectors encoding Myc-epitope-tagged T6BP and the Flag-tagged TRAFs 1–6. Cell extracts were prepared and immunoprecipitated (IP) with anti-Flag monoclonal antibody (F) or control mouse IgG (C). Coprecipitating Myc-T6BP was detected by immunoblotting (IB) analysis with the anti-Myc polyclonal antibody (Top). The amount of TRAFs immunoprecipitated and the expression level of Myc-T6BP in total cell extracts were determined by immunoblotting with anti-Flag polyclonal antibody (Middle) and anti-Myc polyclonal antibody (Bottom). The positions of T6BP and TRAFs are indicated. KD, kilodalton.
N-Terminal Ring Finger and Zinc Finger Regions of TRAF6 Are Required for Its Interaction with T6BP.
TRAF6 contains an N-terminal ring finger domain (amino acids 1–110) followed by five zinc fingers (amino acids 130–273). The C-terminal TRAF domain of TRAF6 can be divided further into TRAF-N (amino acids 274–350) and TRAF-C (amino acids 351–522) domains. To determine which regions of TRAF6 contribute to T6BP binding, various TRAF6 deletion mutants were generated as either GST-tagged proteins or Flag-tagged proteins. The TRAF6 deletion constructs were cotransfected with an expression vector encoding myc-tagged T6BP (amino acids 185–747) into 293 cells, and protein–protein interaction experiments were performed. Deletion of the N-terminal ring finger and zinc fingers of TRAF6 abolishes the association between TRAF6 and T6BP (Fig. 3A). The N-terminal region (amino acids 1–354) of TRAF6, which contains the ring and zinc fingers, was able to bind T6BP, but the C-terminal TRAF domain (amino acids 289–522) of TRAF6 does not bind T6BP (Fig. 3B). These data demonstrate that the interaction between TRAF6 and T6BP requires the N terminus of TRAF6.
Figure 3.
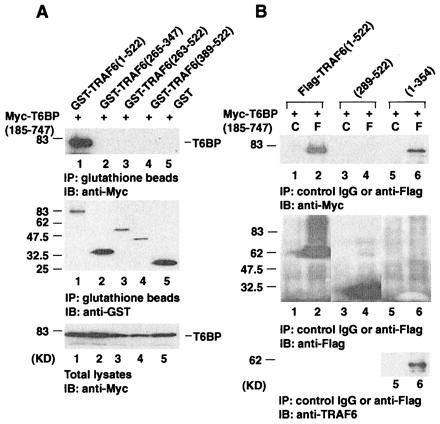
The N-terminal ring finger and zinc finger regions of TRAF6 are required for its interaction with T6BP. (A) 293 cells were transiently transfected with the Myc-epitope tagged T6BP (amino acids 185–747) and the GST-tagged TRAF6 deletion mutants. Cell extracts were prepared and immunoprecipitated (IP) with glutathione beads. Coprecipitating Myc-T6BP was detected by immunoblotting (IB) analysis with the anti-Myc polyclonal antibody (Top). The amounts of GST-TRAF6 deletion mutants immunoprecipitated and the expression levels of Myc-T6BP in total cell extracts were determined by immunoblotting with anti-GST antibody (Middle) and anti-Myc antibody (Bottom). (B) Myc-epitope tagged T6BP (amino acids 185–747) and the Flag-tagged TRAF6 deletion mutants were transiently expressed in 293 cells. Cell extracts were prepared and immunoprecipitated with anti-Flag monoclonal antibody (F) or control mouse IgG (C). Coprecipitating Myc-T6BP was detected by immunoblotting analysis with the anti-Myc polyclonal antibody (Top). The amounts of TRAF6 deletion mutants immunoprecipitated were determined by immunoblotting with anti-Flag polyclonal antibody (Middle). The expression of Flag-TRAF6 (amino acids 1–354) is weak but can be detected with anti-TRAF6 monoclonal antibody (Bottom). KD, kilodalton.
Domains in T6BP That Are Responsible for TRAF6-Binding and Self-Association.
T6BP contains three coiled-coil domains and two helix–loop–helix domains. To determine which regions of T6BP contribute to TRAF6 binding, various T6BP deletion mutants were generated. Flag-tagged TRAF6 and Myc-tagged T6BP deletion mutants were coexpressed in 293 cells, and immunoprecipitation experiments were performed. The results indicate that T6BP has two independent TRAF6-binding domains: either amino acids 1–320 (a domain that includes the first coiled-coil region of T6BP) or amino acids 321–747 (a domain that includes the second and third coiled-coil regions of T6BP) alone can interact with TRAF6 (Fig. 4). T6BP is also capable of self-association as demonstrated by experiments in which Flag-tagged full-length T6BP and Myc-tagged T6BP deletion mutants were coexpressed in 293 cells. The second coiled-coil region of T6BP (amino acids 320–420) is responsible for T6BP self-association (Fig. 5).
Figure 4.

The TRAF6-interacting domains in T6BP. 293 cells were transiently transfected with expression vectors encoding Myc-epitope tagged T6BP deletion mutants and Flag-tagged TRAF6. Cell extracts were prepared and immunoprecipitated (IP) with anti-Flag monoclonal antibody (F) or control mouse IgG (C). Coprecipitating Myc-T6BP deletion mutants were detected by immunoblotting (IB) analysis with the anti-Myc polyclonal antibody (Top). The amounts of TRAF6 immunoprecipitated and the expression level of Myc-T6BP deletion mutants in total cell extracts were determined by immunoblotting with anti-Flag polyclonal antibody (Middle) and anti-Myc antibody (Lower). KD, kilodalton.
Figure 5.
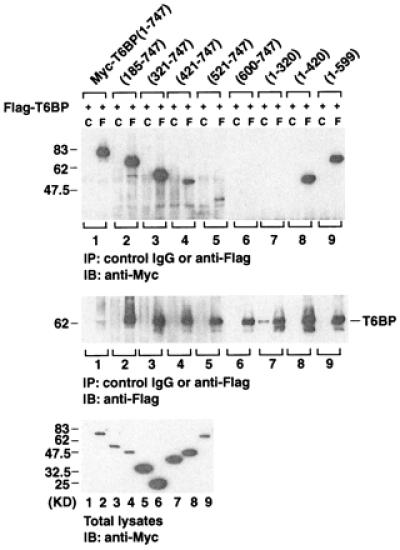
Self-association of T6BP. Myc-epitope tagged T6BP deletion mutants and the Flag-tagged full-length T6BP were coexpressed in 293 cells. Cell extracts were prepared and immunoprecipitated (IP) with anti-Flag monoclonal antibody (F) or control mouse IgG (C). Coprecipitating Myc-T6BP deletion mutants were detected by immunoblotting (IB) analysis with the anti-Myc polyclonal antibody (Top). The amounts of Flag-T6BP immunoprecipitated and the expression level of Myc-T6BP deletion mutants in total cell extracts were determined by immunoblotting with anti-Flag polyclonal antibody (Middle) and anti-Myc antibody (Lower).
IL-1-Inducible Interactions Among TRAF6, T6BP, and IRAK.
To determine whether TRAF6 and T6BP interact endogenously, we used a 293 cell line stably expressing IL-1RI, which responds well to IL-1 stimulation (4). Cells were stimulated with IL-1, lysed, and immunoprecipitated with an anti-T6BP rabbit polyclonal antibody. Endogenous TRAF6 was coprecipitated with T6BP in a time-dependent manner; TRAF6 is readily detectable 5 min after IL-1 stimulation and peaks at 30 min (Fig. 6A Top). IRAK is not present in these T6BP immunoprecipitates (Fig. 6A Bottom).
Figure 6.
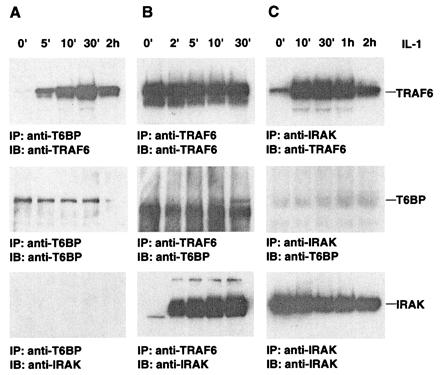
IL-1-inducible interactions among TRAF6, T6BP, and IRAK. 293.IL-1RI cells were stimulated with 10 ng/ml IL-1 for various times. Cell lysates were immunoprecipitated (IP) with an anti-T6BP rabbit polyclonal antibody (A), anti-TRAF6 polyclonal antibody (B), or anti-IRAK polyclonal antibody (C). Coprecipitating endogenous proteins were detected by immunoblotting (IB) with anti-TRAF6 monoclonal antibody (Top), anti-T6BP polyclonal antibody (Middle), or anti-IRAK polyclonal antibody (Bottom). The positions of TRAF6, T6BP, and IRAK are indicated.
To explore the complex formation between TRAF6, T6BP, and IRAK further, the IL-1-stimulated 293.IL-1RI cell lysates were immunoprecipitated with either anti-TRAF6 (Fig. 6B) or anti-IRAK (Fig. 6C) antibodies and immunoblotted with antibodies against TRAF6, T6BP, or IRAK. Although T6BP and IRAK can be recruited to TRAF6 after IL-1 treatment, T6BP and IRAK are not present in the same complex. These results suggest that TRAF6–T6BP and TRAF6–IRAK exist as two mutually exclusive complexes.
IRAK Is Required for the IL-1-Inducible TRAF6–T6BP Complex Formation.
To investigate the role of IRAK in IL-1-induced TRAF6–T6BP complex formation, we used the IRAK-deficient I1A cell line, which was derived from 293.IL-1RI cells through mutagenesis (27). The T6BP–TRAF6 complex is not detectable in IL-1-treated I1A cells (Fig. 7A), indicating that IRAK is required for the IL-1-induced interaction between TRAF6 and T6BP. Interestingly, when I1A cells are transiently transfected with an IRAK expression plasmid, the TRAF6–T6BP interaction was restored (Fig. 7B). However, this interaction was now IL-1-independent, probably because of the constitutive activation of IRAK during transfection.
Figure 7.
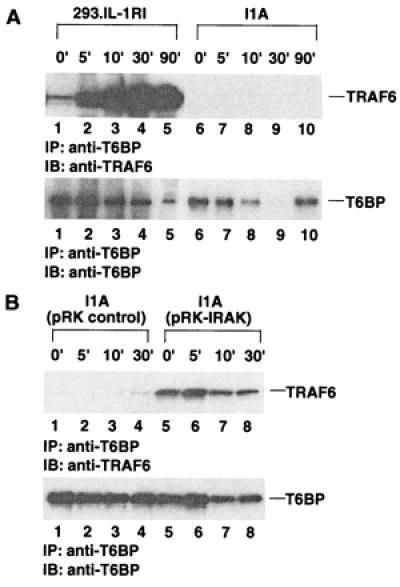
IRAK is required for the IL-1-inducible TRAF6–T6BP complex formation. (A) 293.IL-1RI cells or I1A cells were stimulated with 10 ng/ml IL-1 for various times. Cell lysates were immunoprecipitated (IP) with anti-T6BP antibody and immunoblotted (IB) with anti-TRAF6 monoclonal antibody (Upper). The amounts of T6BP in the immunoprecipitates were determined by immunoblotting with anti-T6BP antibody (Lower). (B) I1A cells were transiently transfected with HA-tagged IRAK in pRK expression vector and stimulated with 10 ng/ml IL-1 for various times. Cell lysates were immunoprecipitated with anti-T6BP antibody and immunoblotted with anti-TRAF6 monoclonal antibody (Upper). The amounts of T6BP in the immunoprecipitates were determined by immunoblotting with anti-T6BP antibody (Lower).
The TRAF6–T6BP Interaction Is IL-1-Specific.
TNF is a proinflammatory cytokine that has similar activities to IL-1. For example, HeLa cells respond equally well to IL-1 and TNF stimulation in terms of NF-κB activation. However, the TRAF6–T6BP complex formation is not detected in HeLa cells after stimulation with TNF (Fig. 8A). Gene-targeting experiments in mice have shown that TRAF6 is also required for CD40, lipopolysaccharide, and IL-17 signaling pathways (5, 20). Therefore, we explored whether the TRAF6–T6BP complex formation occurs after CD40 ligand stimulation. In 293.CD40 cells (a 293 cell line stably expressing Flag-tagged CD40), stimulation with a membrane preparation of CD40 ligand does not result in the inducible TRAF6–T6BP association (Fig. 8B Left), although TRAF6 is recruited to the activated CD40 receptor in a time-dependent manner (Fig. 8B Right).
Figure 8.

The TRAF6–T6BP interaction is IL-1-specific. (A) HeLa cells were stimulated with 10 ng/ml IL-1 or 100 ng/ml TNF for various times. Cell lysates were immunoprecipitated (IP) with anti-T6BP antibody and immunoblotted (IB) with anti-TRAF6 monoclonal antibody (Upper). The amounts of T6BP in the immunoprecipitates were determined by immunoblotting with anti-T6BP antibody (Lower). (B) 293.CD40 cells were stimulated with a membrane preparation of CD40 ligand for various times. Cell lysates were immunoprecipitated with anti-T6BP antibody and immunoblotted with anti-TRAF6 monoclonal antibody (Upper Left). The amounts of T6BP in the immunoprecipitates were determined by immunoblotting with anti-T6BP antibody (Lower Left). The same cell lysates were also immunoprecipitated with anti-Flag antibodies that recognize the Flag-tagged CD40 receptor and were immunoblotted with anti-TRAF6 monoclonal antibody (Upper Right). The amounts of CD40 receptor in the immunoprecipitates were determined by immunoblotting with anti-CD40 antibody (Lower Right).
T6BP Is Not an Activator of NF-κB or JNK Pathways.
As a proinflammatory cytokine, IL-1 initiates a cascade of signaling events leading to the activation of NF-κB and JNK. TRAF6 is required for NF-κB and JNK activation by IL-1. When transiently expressed in 293 cells, TRAF6, but not T6BP, can activate an NF-κB luciferase reporter in a dose-dependent manner (Fig. 9A). To test wether T6BP can activate the JNK pathway, TRAF6 or T6BP was coexpressed with a HA-tagged JNK construct in 293 cells. Although TRAF6 activates HA-JNK in an in vitro kinase assay by using GST-c-Jun as a substrate, T6BP does not (Fig. 9B). In addition, T6BP does not inhibit IL-1-induced NF-κB or JNK activation in cotransfection experiments (data not shown). These results indicate that T6BP may play a role in signaling IL-1 responses distinct from NF-κB or JNK activation.
Figure 9.

T6BP is not an activator of NF-κB or JNK pathways. (A) 293 cells were transiently cotransfected with an E-selectin-luciferase reporter gene plasmid and various amounts (given in micrograms per 35-mm plate) of TRAF6 or T6BP expression plasmids. At 24 h after transfection, luciferase activities were determined and normalized on the basis of β-galactosidase expression. (B) 293 cells were transiently cotransfected with HA-JNK plasmid and various amounts (given in micrograms per 100-mm plate) of TRAF6, T6BP, or MEKK1 expression plasmids. Cell lysates were immunoprecipitated (IP) with anti-HA antibody and incubated with [γ-32P]ATP in the presence of bacterially expressed GST-Jun (amino acids 1–79). The proteins were resolved by SDS/PAGE and analyzed by autoradiography (Upper). The HA-JNK protein expression levels were determined by immunoblotting (IB) with anti-HA antibody (Lower). The positions of GST-Jun (1–79) and HA-JNK are indicated.
Discussion
IL-1 is a proinflammatory cytokine that participates in defense response to environmental challenges by generating fever, activating lymphocytes, and promoting infusion of leukocytes into the sites of injury or infection (32). IL-1 signals through a receptor complex of IL-1RI and the IL-1R accessory protein to generate multiple cellular responses. IL-1 binding to the receptor complex results in the recruitment of the adaptor molecule MyD88 and the serine/threonine kinase IRAK. IRAK becomes autophosphorylated and then interacts with TRAF6, which transduces the IL-1 signal downstream to NF-κB and JNK activation.
In this study, we report the identification of a TRAF6-interacting protein, T6BP. Interestingly, TRAF6 is the only TRAF protein that interacts with T6BP, and this interaction requires the N-terminal ring finger/zinc finger domains of TRAF6. To our knowledge, this is the first case of a TRAF-binding protein that interacts with a single member of the TRAF family specifically through ring finger/zinc finger domains. The ring finger/zinc finger domains of TRAF proteins are thought to be important in transducing signals to downstream targets (1, 4, 11, 14). Like TRAF proteins, T6BP can also self-associate.
Previously, we identified a protein, MIP-T3, that specifically associates with TRAF3 (26). TRAF3 dissociates from the TRAF3–MIP-T3 complex and is recruited to the CD40 receptor on CD40 ligand stimulation (26). Herein, we present another TRAF-binding protein, T6BP, whose interaction with TRAF6 can be induced by IL-1. Identification and characterization of TRAF-binding proteins specific for single members of the TRAF family will be important for understanding the different roles played by individual TRAF proteins.
The association of TRAF6 with T6BP is also different from the interaction of TRAF2 with c-IAP1 (33, 34). In TNF receptor signaling, TRAF2 and c-IAP1 are rapidly recruited to both TNFR1 and TNFR2 signaling complexes in a TNF-dependent manner in untransfected mammalian cells (34). TRAF2 and c-IAP1 are preassociated, and the TRAF2–c-IAP1 complex interacts with TNFR2 via TRAF2 (33, 34), whereas the recruitment of TRAF2 and c-IAP1 to TNFR1 is mediated by TRADD (13, 34). In the case of IL-1 receptor signaling, IRAK, not TRAF6, is recruited to the activated receptors. IRAK becomes highly phosphorylated and then forms a complex with TRAF6 that is not associated with the IL-1 receptor.
Our results suggest that IRAK is required for IL-1-induced TRAF6–T6BP complex formation. Like TRAF6, IRAK has been shown both biochemically and genetically to be essential for NF-κB and JNK activation by IL-1 (35–37). It is tentative to place TRAF6–T6BP complex downstream of TRAF6–IRAK complex. It can be postulated that only “modified” TRAF6, either aggregated or phosphorylated by IRAK, is therefore capable of interacting with T6BP.
Although gene targeting data in mice suggest that TRAF6 is also important in CD40 signaling, CD40 ligand does not induce the TRAF6–T6BP complex formation. These results agree well with the requirement for IRAK in TRAF6–T6BP complex formation. In CD40 signaling, TRAF6 is directly recruited to the activated receptor, bypassing the dependence on IRAK of the IL-1 pathway (11). It will be interesting to investigate whether activation of IL-18 or Toll signaling pathways induces the interaction between TRAF6 and T6BP, because IRAK is implicated in these pathways (38, 39).
TRAF6 is essential for NF-κB and JNK activation by IL-1. However, T6BP neither activates nor inhibits either of these two pathways in transient transfection experiments (Fig. 9 and data not shown). These results provide clues for bifurcation of the NF-κB/JNK pathways and other pathways at the level of TRAF6. Additional efforts will be required to determine the specific functional roles of the T6BP–TRAF6 complex in IL-1 signaling and of T6BP in other pathways.
Acknowledgments
We are indebted to Dr. Zhandan Cao for providing various reagents and valuable advice. We thank Dr. Hsing-Jien Kung for providing the pretransformed HeLa library for two-hybrid screening. We thank Dr. Ralf Schwandner for generously providing the 293.CD40 cell line and CD40 ligand before publication. We thank Ms. Miki Rich for DNA sequencing. We would also like to thank Dr. Holger Wesche and Dr. Guoqing Chen for stimulating discussions.
Abbreviations
- IL-1RI
IL-1 receptor I
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TRAF
TNFR-associated factor
- JNK
c-jun N-terminal kinase
- IRAK
IL-1 receptor-associated kinase
- GST
glutathione S-transferase
- HA
hemagglutinin
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF268075).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170279097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170279097
References
- 1.Rothe M, Wong S C, Henzel W J, Goeddel D V. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 2.Hu H M, O'Rourke K, Boguski M S, Dixit V M. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- 3.Cheng G, Cleary A M, Ye Z S, Hong D I, Lederman S, Baltimore D. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 4.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel D V. Nature (London) 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 5.Lomaga M A, Yeh W C, Sarosi I, Duncan G S, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, Irie S, Reed J C. FEBS Lett. 1995;358:113–118. doi: 10.1016/0014-5793(94)01406-q. [DOI] [PubMed] [Google Scholar]
- 8.Regnier C H, Tomasetto C, Moog-Lutz C, Chenard M P, Wendling C, Basset P, Rio M C. J Biol Chem. 1995;270:25715–25721. doi: 10.1074/jbc.270.43.25715. [DOI] [PubMed] [Google Scholar]
- 9.Ishida T K, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J. Proc Natl Acad Sci USA. 1996;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware C F, Yagita H, Okumura K. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- 11.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, et al. J Biol Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi M, Rothe M, Goeddel D V. J Biol Chem. 1996;271:19935–19942. doi: 10.1074/jbc.271.33.19935. [DOI] [PubMed] [Google Scholar]
- 13.Hsu H, Shu H B, Pan M G, Goeddel D V. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 14.Rothe M, Sarma V, Dixit V M, Goeddel D V. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 15.Song H Y, Regnier C H, Kirschning C J, Goeddel D V, Rothe M. Proc Natl Acad Sci USA. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh W C, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa J L, Ferrick D, Hum B, Iscove N, et al. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen L T, Duncan G S, Mirtsos C, Ng M, Speiser D E, Shahinian A, Marino M W, Mak T W, Ohashi P S, Yeh W C. Immunity. 1999;11:379–389. doi: 10.1016/s1074-7613(00)80113-2. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Cheng G, Baltimore D. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 19.Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, Munechika E, Sakai T, Shirasawa T, Akiba H, et al. Proc Natl Acad Sci USA. 1999;96:9803–9808. doi: 10.1073/pnas.96.17.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwandner R, Yamaguchi K, Cao Z. J Exp Med. 1999;191:1233–1240. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wesche H, Henzel W J, Shillinglaw W, Li S, Cao Z. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway C A., Jr Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 23.Muzio M, Ni J, Feng P, Dixit V M. Science. 1997;278:1612–1615. [Google Scholar]
- 24.Karin M, Delhase M. Semin Immunol. 1999;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 25.Arch R H, Gedrich R W, Thompson C B. Genes Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 26.Ling, L. & Goeddel, D. V. (2000) J. Biol. Chem., in press. [DOI] [PubMed]
- 27.Li X, Commane M, Burns C, Vithalani K, Cao Z, Stark G R. Mol Cell Biol. 1999;19:4643–4652. doi: 10.1128/mcb.19.7.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling L, Cao Z, Goeddel D V. Proc Natl Acad Sci USA. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z G, Hsu H, Goeddel D V, Karin M. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 30.De Valck D, Jin D Y, Heyninck K, Van de Craen M, Contreras R, Fiers W, Jeang K T, Beyaert R. Oncogene. 1999;18:4182–4190. doi: 10.1038/sj.onc.1202787. [DOI] [PubMed] [Google Scholar]
- 31.Uyeda A, Simo Wydisdtuti M, Nishimune H, Kiyosue K, Kasai M, Taguchi T. Neurosci Lett. 2000;284:61–64. doi: 10.1016/s0304-3940(00)00959-9. [DOI] [PubMed] [Google Scholar]
- 32.Dinarello C A. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 33.Rothe M, Pan M G, Henzel W J, Ayres T M, Goeddel D V. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 34.Shu H B, Takeuchi M, Goeddel D V. Proc Natl Acad Sci USA. 1996;93:13973–13978. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao Z, Henzel W J, Gao X. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 36.Kanakaraj P, Ngo K, Wu Y, Angulo A, Ghazal P, Harris C A, Siekierka J J, Peterson P A, Fung-Leung W P. J Exp Med. 1999;189:1129–1138. doi: 10.1084/jem.189.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas J A, Allen J L, Tsen M, Dubnicoff T, Danao J, Liao X C, Cao Z, Wasserman S A. J Immunol. 1999;163:978–984. [PubMed] [Google Scholar]
- 38.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 39.Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. J Exp Med. 1998;187:2097–2101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]