

Abstract
Pma1-D378N is a misfolded plasma membrane protein in yeast that is prevented from delivery to the cell surface and targeted instead for ER-associated degradation (ERAD). Degradation of Pma1-D378N is dependent on the ubiquitin ligase Doa10 and the ubiquitin chaperone Cdc48. Recognition of Pma1-D378N by the ERAD pathway is dependent on Eps1, a transmembrane member of the protein disulfide isomerase (PDI) oxidoreductase family. Eps1 has two thioredoxin-like domains containing a CPHC and a CDKC active site. Although Eps1 interaction with wild-type Pma1 was not detected, Eps1 co-immunoprecipitates with Pma1-D378N. Eps1 interaction with Pma1-D378N requires the CPHC motif, although both thioredoxin-like domains appear to cooperate in substrate recognition. In the absence of the native transmembrane domain and cytoplasmic tail of Eps1, degradation of Pma1-D378N is slowed, suggesting that Eps1 facilitates presentation of substrate to membrane-bound components of the degradation machinery. Genetic interactions with other mutants of the ERAD machinery and induction of the unfolded protein response in eps1Δ cells support a general role for Eps1 as a recognition component of the ERAD pathway.
Keywords: endoplasmic reticulum/ER-associated degradation/plasma membrane ATPase/protein disulfide isomerase/unfolded protein response
Introduction
A quality control mechanism operates in the endoplasmic reticulum (ER) to monitor and assist the proper folding and assembly of newly synthesized proteins (Ellgard et al., 1999). At the same time, conformationally aberrant proteins detected by ER quality control elicit two major responses: the unfolded protein response (UPR) and ER-associated degradation (ERAD). UPR is a signal transduction pathway leading from the ER sensor Ire1 to transcriptional activation of a large number of proteins, including folding enzymes and chaperones, in order to increase the folding potential within the ER (Spear and Ng, 2001). ERAD is a route by which misfolded proteins are recognized, prevented from ER export, and delivered for retrotranslocation from ER to cytosol and degradation via the ubiquitin–proteasome pathway (Bonifacino and Weissman, 1998; Tsai and Rapoport, 2002).
A critical activity in ER quality control is to discriminate between a protein in the process of folding and a substrate for ERAD (Fewell et al., 2001; Tsai et al., 2002). Given the conformational diversity of newly made proteins in the ER, it is not surprising that there are multiple mechanisms for recognition of misfolded proteins. Some ERAD substrates are recognized by their glycosylation state while a number of chaperones, including the lumenal Hsp70, BiP, have been implicated in directing misfolded proteins to the ERAD pathway. There is accumulating evidence that the multifunctional protein disulfide isomerase (PDI), with oxidoreductase activity as well as chaperone activity, also participates in recognition of some ERAD substrates (Gillece et al., 1999; Sevier and Kaiser, 2002; Tsai et al., 2002).
A membrane substrate of the ERAD pathway, Pma1-D378N, has been characterized previously. In Saccharomyces cerevisiae, Pma1 is a plasma membrane H+-ATPase that fulfills an essential function of generating the membrane potential and regulating intracellular pH by pumping protons out of the cell. Electron crystallography evidence supports hydropathy predictions that Pma1 is a highly asymmetric molecule, with a large cytoplasmic domain, and the extracytoplasmic part consists only of small stretches connecting the 10 transmembrane segments (Kuhlbrandt et al., 2002). Newly synthesized Pma1 assembles as an oligomer in the ER (Lee et al., 2002), and is delivered to the cell surface via the secretory pathway (Chang and Slayman, 1991). Pma1 acquires post-translational modification by phosphorylation at Ser and Thr residues but is not glycosylated (Chang and Slayman, 1991), nor does it appear to have disulfide bridges (Petrov and Slayman, 1995). The Pma1-D378N mutant is a misfolded protein [as assessed by limited trypsinolysis (Nakamoto et al., 1998)], prevented from ER export and degraded in a manner dependent on Ubc6 and Ubc7 (Wang and Chang, 1999). The pma1-D378N allele has a dominant-negative effect on cell growth because it oligomerizes with wild-type Pma1 such that both are targeted for ERAD (Wang and Chang, 2002).
EPS1 was identified in a genetic screen for mutants that suppress the dominant-negative growth phenotype of pma1-D378N (Wang and Chang, 1999). In eps1Δ cells, Pma1-D378N escapes ER retention and degradation, and moves to the cell surface along with wild-type Pma1. On the basis of this phenotype, Eps1 has been proposed to play an essential role in recognition of Pma1-D378N for delivery to the ERAD pathway. Eps1 belongs to the protein disulfide isomerase (PDI) family, whose members have one or two conserved thioredoxin-like domains containing a Cys-X-X-Cys active site motif (Sevier and Kaiser, 2002). Depending on the redox state of the cysteine pair in the active site, thiol-disulfide oxidoreductases are able to catalyze formation, reduction or rearrangement (isomerization) of disulfide bonds. There are five PDI family members in yeast: Pdi1, Mpd1, Mpd2, Eug1 and Eps1. Of these, all are ER lumenal proteins except for Eps1, which is a transmembrane protein (Wang and Chang, 1999), and only Pdi1 is essential. Overexpression of any of the non-essential family members can suppress lethality in the absence of Pdi1 (Norgaard et al., 2001).
In this report, we have examined the molecular mechanism by which Eps1 targets Pma1-D378N for ERAD. We show that mutant Pma1 co-immunoprecipitates with Eps1 but wild-type Pma1 does not. The CPHC motif within the thioredoxin-like domain of Eps1 is essential for interaction with mutant Pma1, and a second CDKC motif cooperates in recognition of substrate. Combined genetic and biochemical evidence suggest a general role for Eps1 as a recognition component of the ERAD machinery.
Results
Structure and function of Eps1
Of the PDI family members in yeast, Eps1 is unique in being a type I transmembrane protein with its thioredoxin domain residing in the ER lumen. Because eps1Δ is a suppressor of pma1-D378N (Wang and Chang, 1999), we tested which structural features are important for Eps1 function by assaying cell growth in the presence of Pma1-D378N. eps1Δ cells bearing pGAL-pma1-D378N grow on galactose (vector, Figure 1A); however, introduction of wild-type EPS1 into these cells reverses the suppression and the cells cannot grow. No difference was detectable in function of EPS1 tagged at either the N- or C-terminus with HA or myc epitopes, respectively, in comparison with wild type (not shown). To determine whether the membrane anchor of Eps1 is required for its ability to direct Pma1-D378N into the ERAD pathway, two constructs were tested in which the transmembrane domain and cytoplasmic tail of Eps1 were deleted and replaced with an HDEL ER retention signal, or the transmembrane domain and tail of Eps1 were replaced with that of the endogenous ER membrane protein Wbp1 (Gaynor et al., 1994). Both of these Eps1 mutants are expressed at levels similar to that of wild-type Eps1, and are ER-localized (not shown). Figure 1A shows that Eps1, with its transmembrane domain either deleted or replaced, acts the same as wild-type Eps1 in preventing growth of cells expressing Pma1-D378N. In contrast, mutation to serine of both cysteines (Cys60 and Cys63) of the CPHC motif within the thioredoxin domain results in complete loss of function of Eps1 and permits growth in the presence of Pma1-D378N (compare vector with SPHS; Figure 1A). Strikingly, the CPHS mutant is nearly as functional as wild-type Eps1. From these results, we can deduce a requirement for the first cysteine of the CPHC motif (Cys60). Moreover, the Eps1–CPHS mutant is functional even in the absence of three other members of the PDI family: Mpd1, Mpd2 and Eug1 (not shown).

Fig. 1. Structure–function analysis of Eps1. Cells were spotted on medium in serial 10-fold dilutions and incubated at 30°C. (A) Suppression of pma1-D378N by eps1Δ. eps1Δ cells (WQY1) bearing pGAL-pma1-D378N (pRN409U/L) were co-transformed with vector, wild-type EPS1 (pWQ53), eps1–HDEL (pWQ57), eps1–wbp1 (pWQ56), eps1–CPHS (pWQ63), eps1–SPHS (pWQ65) or high copy EUG1 (pCT44). All EPS1 plasmids are centromeric. Cells were spotted on synthetic complete medium with 2% galactose or 2% glucose. (B) Suppression of pma1-D378N. Wild-type cells (F1105) bearing pGAL-pma1-D378N were co-transformed with Hac1i (pDN390), high copy EUG1 (pCT44) or vector (–). eug1Δ (M4426), doa10 (MHY1631), ubc6 ubc7 (MHY552), eps1Δ ire1Δ (WQX1-1A) and ire1Δ (MN5) cells are compared with eps1Δ (WQY1), all bearing pGAL-pma1-D378N. (C) Sensitivity to canavanine and cadmium. eps1Δ cells (WQY1) bearing vector, wild-type EPS1 (pWQ53), eps1–HDEL (pWQ57), eps1–wbp1 (pWQ56), eps1–CPHS (pWQ63) or eps1–SPHS (pWQ65). All plasmids are centromeric. Cells were spotted on minimal medium with canavanine (150 µg/ml) or cadmium (5 µM), or synthetic complete medium.
The PDI family member Eug1 has CXXS motifs in two thioredoxin-like domains (Tachibana and Stevens, 1992). In the absence of EPS1, overexpression of EUG1 can partially replace Eps1 function so that growth in the presence of Pma1-D378N is substantially impaired (compare vector and EUG1; Figure 1A). In the presence of EPS1, high copy EUG1 has no discernible effect (Figure 1B, left panel). In eug1Δ (EPS1+) cells, there is some growth of cells expressing Pma1-D378N (although growth is slight in comparison to that seen in eps1Δ cells), suggesting the possibility that Eug1 may assist the function of Eps1.
Accumulation of misfolded protein in the ER activates the UPR, and UPR induction is required for ERAD of some protein substrates (Casagrande et al., 2000). Indeed, UPR is activated in the absence of Eps1 (see below). However, in eps1Δ cells, UPR induction is not necessary for suppression of pma1-D378N: eps1Δ ire1Δ cells [which cannot induce UPR (Cox and Walter, 1996)] still grow on galactose in the presence of pGAL-pma1-D378N (Figure 1B). Conversely, to address whether UPR induction is sufficient to suppress pma1-D378N, cells were transformed with a plasmid expressing intron-less Hac1 (Hac1i) mRNA, resulting in constitutive induction of UPR (Cox and Walter, 1996). Figure 1B shows that Hac1i does not allow cells to grow in the presence of Pma1-D378N, suggesting that UPR induction does not result in release of Pma1-D378N and associated wild-type Pma1 from the ER. Taken together, the results in Figure 1A and B suggest that ER retention and ERAD of Pma1-D378N require the first cysteine (Cys60) of the CPHC motif of Eps1 but that UPR induction is not necessary.
To determine whether Eps1 has a general role in ER quality control, growth of eps1Δ cells in the presence of canavanine and cadmium was tested (Figure 1C). Canavanine, an arginine analog, and heavy metals such as cadmium cause protein misfolding and induce UPR (Kaufman, 1999). Indeed, a number of mutants defective in ERAD, as well as other ubiquitin-dependent pathways, display sensitivity to canavanine and cadmium (Heinemeyer et al., 1993; Jungmann et al., 1993; Swanson et al., 2001). Figure 1C shows that eps1Δ cells grow poorly in comparison to EPS1+ cells in the presence of canavanine and cadmium, reflecting increased sensitivity to accumulation of misfolded proteins. Strikingly, eps1 mutants in the transmembrane domain as well as the thioredoxin motif do not fully complement this phenotype of eps1Δ cells (Figure 1C), although mutants in the transmembrane domain act much like wild-type EPS1 in reversing suppression of pma1-D378N (Figure 1A). Thus, the transmembrane domain does play an important role in Eps1 function (also see below).
Components required for ERAD of Pma1-D378N
In order to understand Pma1-D378N delivery to the ERAD pathway better, we determined which ERAD components are required for its degradation. Pma1-D378N stability was assessed by pulse–chase experiments in several mutants defective in ERAD. The AAA ATPase, Cdc48, has been suggested to act as a ubiquitin chaperone facilitating interaction between ubiquitylated ERAD substrate and the 26S proteasome (Rape et al., 2001); cdc48 mutants are defective in dislocation (Ye et al., 2001; Jarosch et al., 2002). As shown in Figure 2A, Pma1-D378N is degraded in wild-type cells; in cdc48-3 cells at the restrictive temperature, degradation of Pma1-D378N is significantly slowed, consistent with the observed ubiquitylation of Pma1-D378N (Wang and Chang, 2002). Consistent with Cdc48 being in a complex with Npl4 and Ufd1 (Bays and Hampton, 2002), Pma1-D378N is also stabilized in npl4 mutants (not shown). Two ER-associated E3 enzymes or ubiquitin ligases, Hrd1/Der3 and Doa10, have been reported to participate in ERAD of different substrates (Bordallo et al., 1998; Bays et al., 2001; Swanson et al., 2001). Figure 2A shows that Pma1-D378N is significantly stabilized in doa10Δ cells, and a slight, but reproducible, stabilization was also observed in hrd1/der3Δ cells. These data indicate that, in contrast to several other membrane protein substrates (Fewell et al., 2001), Pma1-D378N becomes ubiquitylated primarily via Doa10, although it can be recognized to a lesser extent by Hrd1. Importantly, doa10 cells do not suppress pma1-D378N, although Pma1-D378N is stabilized (Figure 1B). Similarly, Pma1-D378N is stabilized in ubc6 ubc7 cells (Wang and Chang, 1999), defective in ER-associated ubiquitin-conjugating enzymes, and yet these cells cannot grow (Figure 1B), suggesting that ER export and plasma membrane delivery are not merely consequences of stabilization in the ER.
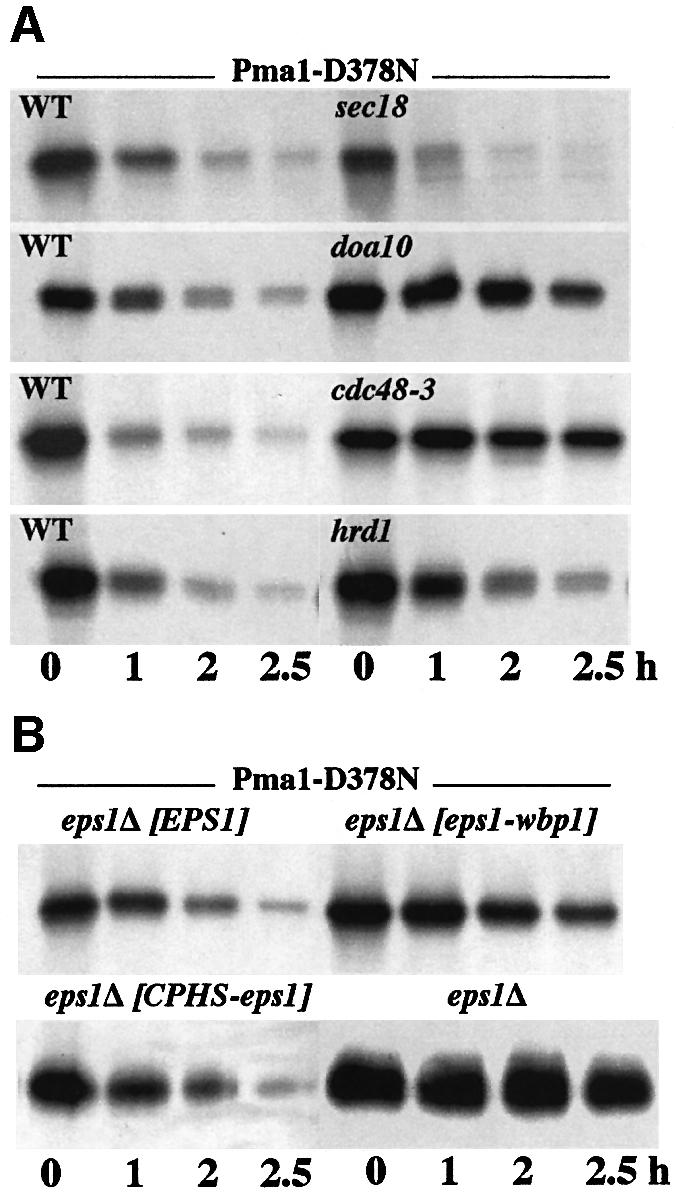
Fig. 2. Requirements for degradation of Pma1-D378N. Synthesis of mutant Pma1 was induced as described in Materials and methods. Cells were radiolabeled with Expre35S35S for 10 min at 25°C and chased in the presence of 10 mM methionine and cysteine for various times. Tagged Pma1 was immunoprecipitated and analyzed by SDS–PAGE and fluorography. (A) Degradation of Pma1-D378N does not require ER export but is dependent on Doa10 and Cdc48. Wild-type (F1105) and sec18 (ACX102-3A), doa10 (MHY1631) and hrd1 (YWO 0431) mutants were transformed with pMET-HA-pma1-D378N (pWQ12). cdc48-3 cells (RSY1181) were transformed with pGAL-myc-pma1-D378N (pWQ2). sec18 and cdc48-3 cells were shifted to 37°C 10 min prior to pulse-labeling at 37°C. (B) The role of Eps1. eps1Δ (WQY1) cells were transformed with pGAL-myc-pma1-D378N (pWQ2) alone or with wild-type EPS1 (pWQ53), eps1–wbp1 (pWQ56) or CPHS–eps1 (pWQ63).
The effect of ER export block on Pma1-D378N degradation was tested because recent work has suggested a pathway for degradation of some substrates involving transport to the Golgi prior to ERAD (Vashist et al., 2001; Haynes et al., 2002). Degradation of Pma1-D378N is as efficient in sec18 cells at the restrictive temperature as in SEC18+ cells (Figure 2A). Thus, ER export is not required for ERAD of Pma1-D378N, consistent with observations thus far that Golgi delivery is required only for certain soluble glycosylated substrates (Vashist et al., 2001; Haynes et al., 2002).
Although loss of its transmembrane domain and cytoplasmic tail does not affect the ability of Eps1 to prevent growth of cells expressing Pma1-D378N (Figure 1A), pulse–chase was used as a more sensitive assay to address the role of the Eps1 transmembrane domain and tail. Because recognition is followed by delivery to the translocon and ubiquitylation of the substrate (Bonifacino and Weissman, 1998; Tsai et al., 2002), it is possible that during ERAD of Pma1-D378N, the transmembrane domain and/or tail of Eps1 facilitates substrate transfer to downstream components of the ERAD machinery. To test this hypothesis, eps1Δ cells were transformed with plasmids bearing pma1-D378N and either wild-type EPS1 or the eps1–wbp1 chimera (in which the transmembrane domain and cytoplasmic tail of Eps1 were replaced with that of Wbp1), and the rate of degradation of mutant Pma1 was examined. Figure 2B shows that Pma1-D378N degradation is significantly slowed in cells expressing Eps1–Wbp1, indicating that the transmembrane domain and tail of Eps1 do indeed facilitate substrate degradation.
Pulse–chase analysis of Pma1-D378N degradation in the presence of the CPHS-Eps1 mutant shows efficient degradation of mutant Pma1 (Figure 2B). These data confirm the growth assay of Figure 1 indicating that the CPHS-Eps1 is able to target Pma1-D378N for ERAD. In contrast, Pma1-D378N is stabilized in the absence of Eps1 (Figure 2B), in agreement with our previous report (Wang and Chang, 1999).
Interaction between Pma1-D378N and Eps1
The simplest model to explain the role of Eps1 in ER retention and ERAD of Pma1-D378N is direct interaction between Eps1 and its substrate. To test this hypothesis, co-immunoprecipitation (co-IP) experiments were performed with HA-tagged Eps1 and either myc-tagged wild-type Pma1 or myc-Pma1-D378N. After induction of myc-Pma1, cells were pulse-labeled with [35S]cysteine and methionine, chased for various times and lysed. Anti-myc IP under denaturing conditions reveals that, after 30 min chase, less Pma1-D378N is recovered in comparison to wild-type Pma1 (Figure 3A), consistent with degradation of mutant Pma1. Strikingly, in cells expressing Pma1-D378N, anti-HA IP of Eps1 under non-denaturing conditions results in an associated 100 kDa protein (Figure 3A). By sequential IP with anti-HA followed by anti-myc, the 100 kDa protein was confirmed as mutant Pma1 (see Figure 4A, lane 6). In contrast, wild-type Pma1 is not co-immunoprecipitated with HA-Eps1 (Figure 3A). Controls in Figure 3B suggest that interaction between Eps1 and mutant Pma1 is specific: no band is immunoprecipitated with anti-HA antibody in the absence of HA-Eps1 (lane 2), and co-IP of myc-Pma1-D378N with HA-Eps1 occurs only upon induction of mutant Pma1 synthesis in galactose medium (lane 4) but not without induction in glucose medium (lane 3).

Fig. 3. Interaction between Eps1 and Pma1-D378N. Cells were induced to express Pma1 by shifting to galactose, as described in Materials and methods. Cells were pulse-labeled with Expre35S35S for 5 min and chased for various times. After cell lysis, IPs of HA-Eps1 and myc-Pma1 were under non-denaturing and denaturing conditions, respectively. IPs were analyzed by SDS–PAGE and fluorography. (A) Time course of interaction. eps1Δ cells (WQY1) were transformed with plasmids bearing HA-EPS1 (pWQ51) and either wild-type GAL-myc-PMA1 (pWQ3) or GAL-myc-pma1-D378N (pWQ4). (B) Specificity of co-IP. eps1Δ cells with only GAL-myc-pma1-D378N were induced to express mutant Pma1 (lanes 1 and 2). eps1Δ cells with both HA-EPS1 and GAL-myc-pma1-D378N were maintained in glucose (–, lane 3), or shifted to galactose medium (+, lane 4). Denaturing IP with anti-myc and non-denaturing with anti-HA. (C) Interaction is prolonged by inhibition of ERAD. doa10Δ cells (MHY1631) bearing plasmids with HA-EPS1 and GAL-myc-pma1-D378N.

Fig. 4. Interaction with Pma1-D378N requires the CPHC motif of Eps1. (A) Interaction of Pma1-D378N with mutant Eps1. eps1Δ (WQY1) cells bearing plasmids with GAL-PMA1 (pWQ3) and wild-type EPS1 (pWQ51) (lanes 1 and 2); eps1Δ cells bearing plasmids with GAL-pma1-D378N (pWQ4) and wild-type EPS1, eps1–SPHS (pWQ66), or eps1–CPHS (pWQ62) (lanes 3–6). Cells were induced to express Pma1, pulse-labeled for 5 min, chased for 5 min and lysed for IP under non-denaturing conditions. Upper: denaturing IP with anti-myc. Lower: lane 1, denaturing IP with anti-myc; lanes 2–5, co-IP with anti-HA; lane 6, sequential IP with anti-myc after co-IP with HA-Eps1. (B) Eps1 has a disulfide bridge. eps1Δ cells bearing wild-type EPS1 or eps1–SPHS were labeled with Expre35S35S for 1 h. Cells were lysed, HA-Eps1 was immunoprecipitated, and analyzed by reducing and non-reducing SDS–PAGE. Double arrows indicate decreased electrophoretic mobility of Eps1 under reducing conditions. (C) Eps1 is in a disulfide-linked complex. eps1Δ bearing pGAL-pma1-D378N and either wild-type EPS1 or eps1–CPHS were grown in glucose medium (– gal) or shifted to galactose medium to induce Pma1-D378N synthesis (+ gal). Lysate was prepared in the presence of 5 mM NEM, resolved by reducing and non-reducing SDS–PAGE, and HA-Eps1 was detected by western blotting with anti-HA. Arrowhead indicates disulfide-linked complex. (D) Co-IPs of Pma1-D378N and Eps1. eps1Δ cells bearing plasmids with GAL-myc-pma1-D378N and wild-type HA-EPS1 or HA-eps1–CPHS were induced to express mutant Pma1 or not. NEM (5 mM) was included in the lysis buffer. Non-denaturing IPs with anti-HA were run on reducing and non-reducing gels and western blotted with anti-HA and anti-myc antibodies. Asterisks indicate high molecular weight bands distinct from the Eps1-containing disulfide-linked complex.
A time course of interaction between Eps1 and Pma1-D378N is shown in Figure 3A. Maximal interaction with newly synthesized Pma1-D378N (arrow) was observed at 5 min chase and declines during chase. [The apparent Mr of Eps1 increases with chase (Figure 3A), reflecting its increasing glycosylation (not shown).] To test whether the time course of association is affected by downstream events in the ERAD pathway, co-IP experiments were performed in doa10Δ cells in which Pma1-D378N degradation is inhibited (Figures 2 and 3C). Figure 3C shows that there is prolonged association between Pma1-D378N and Eps1 in doa10Δ cells, in contrast to DOA10+ cells in which the interaction was observed to diminish substantially by 1 h chase.
Because the CPHC motif is essential for Eps1 function (Figure 1), the CPHS and SPHS mutants were tested for interaction with Pma1-D378N (Figure 4A). Cells expressing mutant Pma1-D378N and wild-type HA-tagged Eps1 or the tagged CPHS or SPHS mutants were pulse-labeled for 5 min and chased for 5 min. After cell lysis, HA-Eps1 was immunoprecipitated under non-denaturing conditions. Figure 4A shows that the Eps1–SPHS mutant (lane 5) fails to associate with Pma1-D378N, but the Eps1–CPHS mutant (lane 4) is competent for interaction. These interactions are consistent with wild-type Eps1 and Eps1–CPHS being functional and able to reverse the suppression of pma1-D378N in eps1Δ cells, but the Eps1–SPHS mutant is not functional (compare Figures 1 and 4A). These data point to the importance of Cys60 in the thioredoxin-like domain of Eps1 in its interaction with its substrate, Pma1-D378N. Similarly, the first cysteine of the active site is required for the activity of PDI (Laboissiere et al., 1995).
There are nine cysteines in Eps1, all in the predicted lumenal domain. Analysis by SDS–PAGE revealed increased electrophoretic mobility of Eps1 under non-reducing conditions in comparison with that seen under reducing conditions (Figure 4B, arrows), indicating that Eps1 has at least one stable intramolecular disulfide bridge. Nevertheless, the mobility of the Eps1–SPHS mutant does not differ from that of wild-type Eps1 (Figure 4B), indicating that the cysteines of the CPHC motif do not participate in a stable intramolecular disulfide linkage.
Analysis of Eps1 under non-reducing conditions revealed a high molecular weight band of ∼185 kDa detected by western blotting (Figure 4C, arrowhead). The accumulated band was observed with the CPHS mutant but not wild-type Eps1 (Figure 4C, lanes 2 and 1) or the SPHS mutant (not shown), and the band is not present under reducing conditions (lane 8). These data suggest the possibility that Eps1 engages in thiol–disulfide exchange reactions and the band reflects Eps1 participation in a mixed disulfide intermediate. To test whether Pma1 is present in the disulfide-linked complex, HA-Eps1 was detected under non-reducing conditions by western blotting following induction of wild-type or Pma1-D378N synthesis. As shown in Figure 4C, a decrease in level of disulfide-linked complex was observed when Pma1-D378N synthesis was induced by shifting to galactose (lane 6), in comparison with that seen in the absence of induction (lane 2) or upon expression of wild-type Pma1 (lane 4). These data suggest that the CPHS-Eps1 is stabilized in a disulfide-linked complex and mutant Pma1 promotes Eps1 loss from the complex. In support of this idea, Pma1-D378N was not detected in the 185 kDa complex: co-IPs of HA-Eps1 and myc-Pma1-D378N were run on non-reducing gels and blotted with anti-HA and anti-myc antibodies (Figure 4D). Minor high molecular weight bands distinct from the 185 kDa complex were detected by western blotting with anti-myc antibody (Figure 4D, asterisks). However, these bands are present under both non-reducing and reducing conditions, and are likely a reflection of Pma1’s propensity to aggregate.
The CPHC motif of Eps1 lies within a domain (comprised of the first 100 residues) that has obvious sequence conservation and clear similarity to the thio redoxin fold (Wang and Chang, 1999; Norgaard et al., 2001). A second CXXC sequence (C200DKC) is present in a region (approximately spanning residues 150–290) with no obvious sequence conservation. Nevertheless, several-fold recognition servers predict with high confidence that this region has similarity to the disulfide isomerase/thioredoxin family (Karplus et al., 1998; Jones, 1999; Kelley et al., 2000; Shi et al., 2001). It therefore appears that Eps1 has two thioredoxin-like domains each with a CXXC motif. To determine whether the CDKC motif acts in collaboration with the CPHC motif, both cysteines of the CDKC motif were changed to serines. Figure 5 shows that Eps1 with triple CPHS, SDKS changes is not able to reverse the suppression of pma1-D378N by eps1Δ cells, in contrast to the CPHS or the SDKS mutants. The CPHS, SDKS changes do not change the electrophoretic mobility of Eps1 under reducing and non-reducing conditions (not shown), indicating that the cysteines of the two CXXC motifs do not participate in intramolecular disulfide bridging. These results suggest that the CDKC motif assists the CPHC motif in recognition of Pma1-D378N.

Fig. 5. The CDKC motif cooperates with the CPHC motif. eps1Δ (WQY1) cells were transformed with plasmids with GAL-pma1-D378N (pRN409U) and wild-type EPS1 (pWQ51), eps1–CPHS (pWQ62), eps1–CPHS, SDKS (pWQ70) and eps1–SDKS (pWQ71). Cells were spotted on synthetic complete medium with glucose or galactose in serial 10-fold dilutions and incubated at 30°C.
A general role for Eps1 in the ERAD pathway
To address whether Eps1 acts as a substrate-specific chaperone or plays a broader role in substrate recognition in ERAD, several experimental approaches were taken. Figure 6A shows genetic interaction between EPS1 and components of the ERAD pathway. Like other mutants defective in ERAD (Heinemeyer et al., 1993), eps1Δ cells are sensitive to canavanine, as are ubc6Δ ubc7Δ mutants (Figure 6A). The eps1Δ ubc6Δ ubc7Δ triple mutant displays severe synthetic sensitivity to canavanine, although growth appears normal in the absence of the arginine analog. Similarly, there is genetic interaction between eps1 and the temperature-sensitive mutant cdc48-3. At the permissive temperature (30°C), cdc48-3 cells do not have a significant growth defect; however, cdc48-3 eps1Δ double mutants have a synthetic growth defect (Figure 6A, bottom right).

Fig. 6. Eps1 plays a general role in ERAD. (A) Genetic interactions between eps1 and cdc48, and eps1 and ubc6 ubc7. Congenic eps1 and cdc48-3 strains (WQX10) were spotted on synthetic complete medium after serial 10-fold dilutions and incubated at 25 and 30°C. Triple eps1 ubc6 ubc7 cells are congenic with ubc6 ubc7 and plated on minimal medium with 150 µg/ml canavanine. (B) Induction of UPR in eps1Δ cells (left). Exponentially growing wild-type (F1105) and eps1Δ (WQY1) cells bearing a UPRE-lacZ reporter construct (pJC104) were lysed and β-galactosidase activity was measured as described (Rose and Botstein, 1983). Cells were treated with tunicamycin (5 µg/ml) and DTT (5 mM) for 1 h at 30°C. UPRE-lacZ activity is expressed in nm/min/mg. The values presented are the mean of 3–5 experiments ± standard error of the mean. Right: EPS1 transcriptional regulation. Cells were grown to mid-log and treated with tunicamycin (+T) and DTT (+D). Total RNA was isolated, 20 µg were loaded per lane, and analyzed by northern blotting using radiolabeled probes from EPS1 and KAR2. ACT1 transcripts were monitored as loading controls. (C) Stabilization of H-2Kb by eps1Δ. eps1Δ cells (WQY1) expressing pGAL-H-2Kb (p210A2) were grown in synthetic complete medium without cysteine and methionine with 2% raffinose. Cells were shifted to medium with 2% galactose for 3 h to induce expression of H-2Kb, and then pulse-labeled for 5 min with Expre35S35S and chased for various times. Cells were lysed for IP with anti-heavy chain antibody. IPs were analyzed by SDS–PAGE and fluorography.
If Eps1 plays a general role in the ERAD pathway, misfolded proteins may accumulate in the absence of Eps1, and this may trigger the unfolded protein response (Friedlander et al., 2000; Spear and Ng, 2001). UPR was examined in eps1Δ cells by using a reporter construct in which a lacZ reporter is fused to the unfolded protein response element (Cox and Walter, 1996). eps1Δ cells bearing the UPRE-lacZ reporter have β-galactosidase levels increased ∼20-fold in comparison to that of wild-type cells, indicating constitutive UPR activation. Constitutive activation of UPR in eps1Δ cells is comparable to that of wild-type cells treated with tunicamycin or DTT, suggesting accumulation of misfolded proteins in the absence of Eps1. Although UPR is activated in eps1Δ cells, northern blot analysis indicates that EPS1 expression is not significantly affected by UPR induction (Figure 6B, right panel). In contrast to KAR2, which is transcriptionally induced upon UPR activation via tunicamycin or DTT treatment (Figure 6B, right lower) (Mori et al., 1992), EPS1 transcript levels remain unaffected (Figure 6B, right upper).
A number of the ERAD substrates established in the literature, including CPY* (Hiller et al., 1996), do not appear to require Eps1 for their recognition (Norgaard et al., 2001; our unpublished results). Perhaps this is not surprising because there are a number of different mechanisms for targeting of a diverse array of ERAD substrates (Fewell et al., 2001; Tsai et al., 2002). Nevertheless, at least one substrate in addition to Pma1-D378N appears stabilized in the absence of Eps1 (Figure 6C). It has been established that the major histocompatibility class I (MHC) heavy chain (H-2Kb) is targeted for ER-associated degradation when expressed in yeast (Casagrande et al., 2000). Pulse–chase analysis indicates that H-2Kb degradation is slowed in eps1Δ cells in comparison with that in EPS1+ cells (Figure 6C), suggesting that Eps1 is important, although not essential, for delivery of H-2Kb to the ERAD pathway. Taken together, genetic and biochemical evidence indicate a general role for Eps1 in ERAD.
Discussion
Pma1-D378N is an ideal substrate for studying ERAD because it inhibits growth by preventing localization of wild-type Pma1 to the cell surface (Wang and Chang, 1999, 2002). By using growth as an assay for cell surface delivery of Pma1, we previously identified eps1 as a mutant that allows escape from ERAD of Pma1-D378N and wild-type Pma1 associated with it (Wang and Chang, 1999). By assaying pma1-D378N suppression, we have now tested the molecular parameters that are involved in recognition of the substrate, Pma1-D378N (Figure 1). Importantly, plasma membrane delivery is not simply a consequence of inhibition of degradation; escape from the ER occurs only when Pma1-D378N is not recognized. Indeed, Pma1-D378N is stabilized in doa10Δ cells (Figure 2) and in ubc6 ubc7 cells (Wang and Chang, 1999), and yet doa10 and ubc6 ubc7 cells are not suppressors of pma1-D378N, i.e. the cells cannot grow when Pma1-D378N is expressed (Figure 1B).
Eps1 is unique among PDI family members in having a membrane anchor. When its transmembrane domain and cytoplasmic tail are replaced by that of the ER resident Wbp1, Eps1 remains competent to deliver Pma1-D378N for ERAD, preventing cell growth (Figure 1A). However, as measured by pulse–chase assay, degradation of Pma1-D378N is slowed in the absence of the native anchor and tail of Eps1 (Figure 2B). It is possible that the transmembrane domain of Eps1 facilitates transfer of ERAD substrate to membrane-bound components of the ERAD machinery. The Eps1 transmembrane domain and tail could mediate direct interaction with downstream ERAD components such as the translocon and ER-associated E2 and E3 enzymes, or they could possibly promote Eps1 localization in a specific ER subdomain. Analogously, during cholera toxin entry into cells by retrotranslocation from ER to cytosol, PDI binding to toxin facilitates membrane association of the complex, presumably as a prelude to transfer through the translocon (Tsai and Rapoport, 2002).
Pma1-D378N was co-immunoprecipitated with Eps1, although no interaction between wild-type Pma1 and Eps1 was detected (Figure 3A); interaction with mutant Pma1 requires the first cysteine (Cys60) of the CPHC motif within the first thioredoxin-like domain of Eps1 (Figures 1A and 4A). By analogy with PDI, which has been proposed to play a role in ERAD as a redox-sensitive chaperone (independent of its enzymatic function as an oxidoreductase) (Gillece et al., 1999; Tsai et al., 2001), it is possible that the redox state of Cys60 of Eps1 regulates its ability to bind Pma1-D378N (via a region distinct from the thioredoxin-like domains).
An alternative model for recognition of Pma1-D378N is that interaction with Eps1 is dependent on formation of a mixed disulfide intermediate involving Cys60. Eps1 appears to engage in a mixed disulfide (with itself or another protein), as reflected by stabilization of the CPHS mutant in a disufide-linked complex, and Pma1-D378N appears to compete with the disulfide-linked complex for Eps1 (Figure 4C). Although a mixed disulfide intermediate between Pma1-D378N and Eps1 was not detected under our experimental conditions (Figure 4D), we cannot exclude this possibility. A direct thiol–disulfide exchange between Eps1 and Pma1-D378N poses a topological problem: the CPHC motif of Eps1 is likely to lie in the ER lumen, whereas no cysteines of native Pma1 are predicted to face the lumen. However, it is possible that Pma1 undergoes folding intermediates in which cysteines are extruded into the lumen, or misfolding of Pma1-D378N leads to lumenal extrusion of a transmembrane cysteine, which could then interact with Eps1. Indeed, thiol-mediated retention is a mechanism by which proteins such as IgM are recognized by ER quality control when they fail to assemble properly (Reddy and Corley, 1998).
If co-IP of Pma1-D378N with Eps1 is mediated by a mixed disulfide with Cys60 of Eps1, the transient nature of the co-IP suggests a free thiol that can break the interaction. The free thiol may come from the second cysteine (Cys63) of the CPHC motif, although our evidence indicates that the CPHS mutant is essentially as functional as wild type (Figures 1A and 4A). It is possible that the Eps1–Pma1-D378N interaction involves other members of the PDI family. It has been demonstrated that PDI family members have overlapping functions: Eug1, with two CXXS motifs, can rescue the lethality of pdi1Δ cells only if Mpd1 and Mpd2 are both present (Norgaard et al., 2001). Nevertheless, recognition of Pma1-D378N by Eps1–CPHS does not appear to depend on Mpd1, Mpd2 and Eug1 (not shown). A free thiol to resolve a mixed disulfide between Eps1 and Pma1-D378N could come from the CDKC motif of Eps1; indeed, the CPHS, SDKS mutant has impaired ability to direct Pma1-D378N to the ERAD pathway (Figure 5), suggesting cooperation between the two thioredoxin-like domains of Eps1.
Several lines of evidence support a general role for Eps1 in ERAD, including genetic interactions between eps1 and other mutants in the ERAD pathway (Figure 6). Whether these genetic interactions indicate physical interactions between Eps1 and other ERAD components awaits further investigation. In the absence of Eps1, induction of UPR implies an increased level of misfolded proteins in the ER, presumably due to inefficient ERAD (Figure 6). Moreover, ERAD of H-2Kb, a transmembrane ERAD substrate like Pma1-D378N, is slowed in eps1Δ cells (Figure 6B). The MHC class I heavy chain is known to have intramolecular disulfides and its ER export is dependent on its assembly with a multicomponent complex, including the oxidoreductase ERp57 (Dick et al., 2002). The possibility is intriguing, in light of thiol-dependent recognition of Pma1-D378N, that delivery of H-2Kb to the ERAD pathway occurs via thiol-mediated interaction with Eps1. Incomplete stabilization of H-2Kb in eps1Δ cells may reflect multiple mechanisms for recognition of this particular substrate.
We have shown that Eps1 plays a role in substrate recognition for ERAD in yeast. It seems possible that Eps1 function is conserved in higher eukaryotes: a database search reveals that transmembrane proteins with thio redoxin-like CXXC active sites are present in all organisms including human.
Materials and methods
Strains and media
Standard yeast media and genetic manipulations were as described (Sherman et al., 1986) and supplemented with 0.2 mM inositol (eps1Δ cells have a wild-type rate of growth in inositol-supplemented media). F1105 is W303 (MATα ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100). WQY1 is MATα ura3-1 leu2-3,112 his3-11 trp1-1 ade2-1 can1-100 eps1Δ::HIS3 (W303 background) (Wang and Chang, 1999). MN5 is MATa his3-11,15 trp1-1 leu2-3,112 ura3-1 ade2-1 can1-100 ire1Δ::LEU2, isogenic with W303 (Nierras and Warner, 1999). RSY1181 (MLY1640) is MATa leu2,3-112 pep4::URA3 cdc48-3 (Latterich et al., 1995). MHY552 is MATα his3Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 ubc6Δ1::HIS3 ubc7::LEU2; MHY1631 is MATα his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 doa10Δ1::HIS3; both strains come from M.Hochstrasser (Yale University, New Haven, CT). ACX102-3A is MATa his3Δ200 lys2Δ201 leu2-3,112 ura3-52, ade2 sec18-1. YWO 0431 is MATα ura3-1 his3-11,15 leu2-3,112 trp1-1 ade2-1ochre can1-100 prc1-1 hrd1/der3::HIS3, provided by Dieter Wolf (Universität Stuttgart, Germany). M4426 is MATα ura3-1 leu2-3,112 his3-11,15 trp1-1 ade2-1 can1-100 eug1Δ (Norgaard et al., 2001). WQX1 is a cross between MN5 and WQY1 to generate an eps1Δ::HIS3 ire1Δ::LEU2 double mutant. WQX10 is a cross between RSY1181 and WQY1 to generate double mutant cdc48-3 eps1Δ::HIS3. WQX5 is a cross to produce a ubc6 ubc7 eps1 triple mutant; before crossing with eps1::LEU2, MHY552 was first transformed with the marker swap plasmid pLT11 (Cross, 1997) to replace the LEU2 marker with a TRP1 marker.
Molecular biology
Plasmids used in this study are listed in Table I. pWQ3 and pWQ4 were constructed by replacing a 3.8 kb HindIII–SalI fragment from pRN409U (Nakamoto et al., 1998) with that from pWQ2 (Wang and Chang, 1999). Full-length EPS1 was cloned as a 7 kb fragment generated by PCR using genomic DNA as template and the oligos AGTCGACCCTCGGT GAGCACCCCAC (no. 118 to introduce a SalI site ∼1 kb 5′ of the EPS1 ATG) and GCCAGAAGATCTGGGATC (no. 128). This fragment was cloned into pGEM-T Easy to generate pWQ24. A triple HA epitope tag was introduced at the N-terminus of Eps1 (between E31 and G32) by site-directed mutagenesis after first using the oligo CTCTGGA AAGCCTCTAGACTCTGGTGGTTC (no. 175) to introduce an XbaI site using pWQ25 as template. For pWQ25, a 3.1 kb SalI–SacI fragment of EPS1 was generated by PCR, cloned into pGEM-TEasy, and then cloned using ApaI–SacI sites into pRS315 (Sikorski and Hieter, 1989). A full-length HA-tagged EPS1 construct was made by replacing the ApaI–XhoI fragment of pWQ24 with the 1.5 kb HA-tagged fragment. pWQ51, pWQ53 and pWQ68 were made by introducing full-length HA-tagged EPS1 into various vector backbones using SalI–NotI sites.
Table I. Plasmids used in this study.
| Name |
Insert |
Description |
Source |
| pRN409U | GAL-pma1-D378N | URA3, CEN | Nakamoto et al. (1998) |
| pWQ2 | GAL-myc-pma1-D378N | LEU2, CEN | Wang and Chang (1999) |
| pWQ12 | MET-HA-pma1-D378N | URA3, CEN | Wang and Chang (1999) |
| pWQ3 | GAL-myc-PMA1 | URA3, CEN | This study |
| pWQ4 | GAL-myc-pma1-D378N | URA3, CEN | This study |
| pWQ51 | HA-EPS1 | LEU2, 2µ | This study |
| pWQ53 | HA-EPS1 | URA3, CEN | This study |
| pWQ62 | HA-eps1-CPHS | LEU2, 2µ | This study |
| pWQ63 | HA-eps1-CPHS | URA3, CEN | This study |
| pWQ65 | myc-eps1-SPHS | LEU2, CEN | This study |
| pWQ66 | HA-eps1-SPHS | LEU2, 2µ | This study |
| pWQ70 | HA-eps1-CPHS, SDKS | LEU2, 2µ | This study |
| pWQ71 | HA-eps1-SDKS | LEU2, 2µ | This study |
| pWQ56 | HA-eps1-WBP1 | URA3, CEN | This study |
| pWQ57 | HA-eps1-HDEL | URA3, CEN | This study |
| PCT44 | GAL-EUG1 | LEU2, 2µ | Tachibana and Stevens (1992) |
| pDN390 | Hac1i | LEU2, CEN | D.Ng (Pennsylvania State University) |
| 210A2 | GAL-H-2Kb | URA3, CEN | Ye et al. (2001) (Pennsylvania State University) |
| pJC104 | UPRE-lacZ | URA3, 2µ | Cox and Walter (1996) |
eps1 mutants were constructed as follows: pWQ56 is a fusion of the N-terminus of Eps1 with the transmembrane domain and cytoplasmic tail of Wbp1. The fusion was made by overlap extension after His648 of Eps1. First, oligos ACCGAAGTGACTCTTCT (no. 166), annealing 1.3 kb downstream of the ATG, and TTCCTAGACATAATCCAC (no. 167), annealing just before the transmembrane domain, were used to generate a 600 bp product using pWQ53 as template. Next, oligos TTCCTAGACATAATCCACTGGGAAATCAGCAATTCT (no. 168) and CAAGCTTGCATGCCTGCA (no. 154) were used to generate an ∼800 bp product with pEG1-KK (Gaynor et al., 1994) bearing WBP1 as template. Finally, oligos 166 and 154 were used as primers to generate a 1.4 kb fusion protein with the 600 and 800 bp products as template. Using HindIII–NotI sites, the fusion was cloned into pWQ53, replacing a 1.2 kb fragment, to generate pWQ56.
pWQ57 contains Eps1 terminating with an HDEL motif instead of its transmembrane domain and cytoplasmic tail. Oligos 118 and CTA CAATTCGTCGTGGTCTAGGAAACTGAGGACATT (no. 177) were used to amplify pWQ53. The product has a SalI site ∼1 kb upstream of the EPS1 start codon and HDEL and stop codon before the transmembrane domain. The product was cloned into pRS316 via pGEM-T Easy at SalI–NotI.
pWQ62, pWQ66, pWQ70 and pWQ71 contain HA-tagged EPS1 with single Cys63Ser (CPHS), double Cys60Ser,Cys63Ser (SPHS), triple Cys63Ser, Cys200Ser, Cys203Ser (CPHS, SDKS) and double Cys200Ser, Cys203Ser (SDKS) mutations, respectively. These constructs were generated by overlap extension. For pWQ62, a 1.2 kb fragment was generated by amplifying pWQ51 using oligos 118 and TAAATGT TTGCTGTGCGGACAGTA (no. 176) to introduce the change. Next, a 1.3 kb fragment was made by amplifying pWQ51 using oligos TACTGTCCGCACAGCAAACATTTAGCA (no. 182) and TCCCAA CTTGCGAGGAGC (no. 183); the fragment terminates ∼30 bp downstream of a unique HindIII site. A 2.5 kb fragment containing Cys63Ser was generated by using oligos 118 and 183 as primers and the 1.2 and 1.3 kb fragments as template. pWQ62 was produced upon replacing SalI–HindIII fragment of pWQ51 with the 2.5 kb PCR fragment. For pWQ66, oligos 118 and TAAATGTTTGCTGTGCGGGGAGTATGGACT (no. 124b), and oligos 186 (AGTCCATACTCCCCGCACAGCAAACAT TTA) and 183 were used to amplify pWQ51. The two products were used as templates for PCR with oligos 118 and 183 to generate a 2.5 kb fragment which was swapped with the SalI–HindIII fragment in pWQ51. For pWQ65, oligo 124b was used for site-directed mutagenesis of pWQ25; the ApaI–XhoI fragment of this construct was swapped with that of pWQ29 [containing full-length myc-tagged Eps1 (Wang and Chang, 1999)]; the SalI–NotI fragment was then subcloned into pRS315. For pWQ70, oligos 118 and 187 (TTCATGGCTTTTGTCACTGTTTT TGAATTC) and 188 (GAATTCAAAAACAGTGACAAAAGCCA TGAA) and 183 were used to amplify pWQ62. The two products were used as templates for PCR with oligos 118 and 183 to generate a 2.4 kb product. The fragment was swapped with the SalI–HindIII fragment of pWQ51 to generate pWQ70. pWQ71 was made in the same way as pWQ70, except that pWQ51 was used as template for the initial PCR reaction. All mutant constructs were confirmed by sequencing.
Northern blotting
Exponentially growing cells were treated with tunicamycin (5 µg/ml) or DTT (5 mM) for 1 h at 30°C. Total RNA was prepared as described, run on agarose gels with formaldehyde, and blotted to Nytran nylon membrane (Nierras and Warner, 1999). 32P-labeled DNA probes for EPS1, KAR2 and ACT1 were prepared by random primer extension. RNA levels were detected by autoradiography.
Metabolic labeling, IP and western blotting
For analysis of cells with GAL1-PMA1 (pWQ3) and GAL1-pma1-D378N (pWQ4), cells were grown overnight at 25°C in minimal medium with glucose or with 2% raffinose and then shifted to medium with galactose for 4 h. For metabolic labeling, cells were resuspended at 1 OD/ml and allowed to rest for 10 min at 25°C before pulse-labeling with Expre35S35S. Temperature-sensitive cdc48-3 and sec18-1 cells were shifted to 37°C 10 min prior to pulse-labeling. An equal volume of synthetic complete medium with 20 mM cysteine and 20 mM methionine was added and cells were chased for various times. For analysis of pma1-D378N under MET25 control, cells were grown in minimal or synthetic complete medium without cysteine and methionine, supplemented with 600 µM methionine. Cells were washed with water and resuspended in medium without methionine for 1 h. Cells were then pulse-labeled for 10 min and chased for various times. For IP and western blotting, cells were lysed by vortexing with glass beads in the presence of a protease inhibitor cocktail including PMSF (Chang and Slayman, 1991). Samples were normalized to acid-precipitable c.p.m. for IP with monoclonal anti-HA (Covance) or polyclonal anti-myc (Santa Cruz) antibodies. Lysate was boiled in 1% SDS and then diluted to 0.1% SDS in RIPA buffer for IP with rabbit anti-heavy chain antibody (gift from Tom Rapoport, Harvard Medical School, Boston, MA). IPs were performed in RIPA buffer or, for IP under non-denaturing conditions, lysate was solubilized in 1% NP-40, 150 mM NaCl, 2 mM EDTA, 10 mM Tris pH 7.5. IPs and lysates were analyzed by SDS–PAGE and fluorography.
Acknowledgments
Acknowledgements
We thank Mark Hochstrasser, Randy Schekman and Martin Latterich, Dieter Wolf, Jakob Winther, Davis Ng, Tom Stevens, Tom Rapoport and Jon Warner for strains, plasmids and antibodies. Thanks to Andras Fiser for help with fold recognition. This work was supported by NIH grant GM 58212.
References
- Bays N.W. and Hampton,R.Y. (2002) Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr. Biol., 12, R366–R371. [DOI] [PubMed] [Google Scholar]
- Bays N.W., Gardner,R.G., Seelig,L.P., Joazeiro,C.A. and Hampton,R.Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol., 3, 24–29. [DOI] [PubMed] [Google Scholar]
- Bonifacino J.S. and Weissman,A.M. (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol., 14, 19–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo J., Plemper,R.K., Finger,A. and Wolf,D.H. (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell, 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casagrande R., Stern,P., Diehn,M., Shamu,C., Osario,M., Zuniga,M., Brown,P.O. and Ploegh,H. (2000) Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell, 5, 729–735. [DOI] [PubMed] [Google Scholar]
- Chang A. and Slayman,C.W. (1991) Maturation of the yeast plasma membrane [H+]ATPase involves phosphorylation during intracellular transport. J. Cell Biol., 115, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.S. and Walter,P. (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell, 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Cross F. (1997) ‘Marker swap’ plasmids: convenient tools for budding yeast molecular genetics. Yeast, 13, 647–653. [DOI] [PubMed] [Google Scholar]
- Dick T.P., Bangia,N., Peaper,D.R. and Cresswell,P. (2002) Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity, 16, 87–98. [DOI] [PubMed] [Google Scholar]
- Ellgard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Fewell S.W., Travers,K.J., Weissman,J.S. and Brodsky,J.L. (2001) The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet., 35, 149–191. [DOI] [PubMed] [Google Scholar]
- Friedlander R., Jarosch,E., Urban,J., Volkwein,C. and Sommer,T. (2000) A regulatory link between ER-associated protein degradation and the unfolded protein response. Nat. Cell Biol., 2, 379–384. [DOI] [PubMed] [Google Scholar]
- Gaynor E.C., te Heesen,S., Graham,T.R., Aebi,M. and Emr,S.D. (1994) Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol., 127, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillece P., Luz,J.M., Lennarz,W.J., de la Cruz,F.J. and Romisch,K. (1999) Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. J. Cell Biol., 147, 1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C.M., Caldwell,S. and Cooper,A.A. (2002) An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol., 158, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer W., Gruhler,A., Mohrle,V., Mahe,Y. and Wolf,D.H. (1993) PRE2, highly homologous to the human major histocompatibility complex-linked RING10 gene, codes for a yeast proteasome subunit necessary for chymotryptic activity and degradation of ubiquitinated proteins. J. Biol. Chem., 268, 5115–5120. [PubMed] [Google Scholar]
- Hiller M.M., Finger,A., Schweiger,M. and Wolf,D.H. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science, 273, 1725–1728. [DOI] [PubMed] [Google Scholar]
- Jarosch E., Taxis,C., Volkwein,C., Bordallo,J., Finley,D., Wolf,D.H. and Sommer,T. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol., 4, 134–139. [DOI] [PubMed] [Google Scholar]
- Jones D.T. (1999) GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol., 287, 797–815. [DOI] [PubMed] [Google Scholar]
- Jungmann J., Reins,H.A., Schobert,C. and Jentsch,S. (1993) Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature, 361, 369–371. [DOI] [PubMed] [Google Scholar]
- Karplus K., Barrett,C. and Hughey,R. (1998) Hidden Markov models for detecting remote protein homologies. Bioinformatics, 14, 846–856. [DOI] [PubMed] [Google Scholar]
- Kaufman R.J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev., 13, 1211–1233. [DOI] [PubMed] [Google Scholar]
- Kelley L.A., MacCallum,R.M. and Sternberg,M.J. (2000) Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol., 299, 499–520. [DOI] [PubMed] [Google Scholar]
- Kuhlbrandt W., Zeelen,J. and Dietrich,J. (2002) Structure, mechanism, and regulation of the Neurospora plasma membrane H+-ATPase. Science, 297, 1692–1696. [DOI] [PubMed] [Google Scholar]
- Laboissiere M.C., Sturley,S.L. and Raines,R.T. (1995) The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J. Biol. Chem., 270, 28006–28009. [DOI] [PubMed] [Google Scholar]
- Latterich M., Frohlich,K.-U. and Schekman,R. (1995) Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell, 82, 885–893. [DOI] [PubMed] [Google Scholar]
- Lee M.C.S., Hamamoto,S. and Schekman,R. (2002) Ceramide biosynthesis is required for the formation of oligomeric H+-ATPase, Pma1p, in the yeast endoplasmic reticulum. J. Biol. Chem., 277, 22395–22401. [DOI] [PubMed] [Google Scholar]
- Mori K., Sant,A., Kohno,K., Normington,K., Gething,M.J. and Sambrook,J.F. (1992) A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J., 11, 2583–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto R.K., Verjovski-Almeida,S., Allen,K.E., Ambesi,A., Rao,R. and Slayman,C.W. (1998) Substitutions of Aspartate 378 in the phosphorylation domain of the yeast PMA1 H+-ATPase disrupt protein folding and biogenesis. J. Biol. Chem., 273, 7338–7344. [DOI] [PubMed] [Google Scholar]
- Nierras C.R. and Warner,J.R. (1999) Protein kinase C enables the regulatory circuit that connects membrane synthesis to ribosome synthesis in Saccharomyces cerevisiae.J. Biol. Chem., 274, 13235–13241. [DOI] [PubMed] [Google Scholar]
- Norgaard P., Westphal,V., Tachibana,C., Alsoe,L., Holst,B. and Winther,J.R. (2001) Functional differences in yeast protein disulfide isomerases. J. Cell Biol., 152, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov V.V. and Slayman,C.W. (1995) Site-directed mutagenesis of the yeast PMA1 H+-ATPase; structural and functional role of cysteine residues. J. Biol. Chem., 270, 28535–28540. [DOI] [PubMed] [Google Scholar]
- Rape M., Hoppe,T., Gorr,I., Kalocay,M., Richly,H. and Jentsch,S. (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48UFD1/NPL4, a ubiquitin-selective chaperone. Cell, 107, 667–677. [DOI] [PubMed] [Google Scholar]
- Reddy P.S. and Corley,R.B. (1998) Assembly, sorting, and exit of oligomeric proteins from the endoplasmic reticulum. BioEssays, 20, 546–554. [DOI] [PubMed] [Google Scholar]
- Rose M. and Botstein,D. (1983) Construction and use of gene fusions to lacZ (β-galactosidase) that are expressed in yeast. Methods Enzymol., 101, 167–180. [DOI] [PubMed] [Google Scholar]
- Sevier C.S. and Kaiser,C.A. (2002) Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol., 3, 836–847. [DOI] [PubMed] [Google Scholar]
- Sherman F., Hicks,J.B. and Fink,G.R. (1986) Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Shi J., Blundell,T.L. and Mizuguchi,K. (2001) FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol., 310, 243–257. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear E. and Ng,D.T.W. (2001) The unfolded protein response: no longer just a special teams player. Traffic, 2, 515–523. [DOI] [PubMed] [Google Scholar]
- Swanson R., Locher,M. and Hochstrasser,M. (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev., 15, 2660–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana C. and Stevens,T.H. (1992) The Yeast EUG1 gene encodes an endoplasmic reticulum protein that is functionally related to protein disulfide isomerase. Mol. Cell. Biol., 12, 4601–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B. and Rapoport,T.A. (2002) Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J. Cell Biol., 159, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B., Rodighiero,C., Lencer,W.I. and Rapoport,T.A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell, 104, 937–948. [DOI] [PubMed] [Google Scholar]
- Tsai B., Ye,Y. and Rapoport,T. (2002) Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol., 3, 246–255. [DOI] [PubMed] [Google Scholar]
- Vashist S., Kim,W., Belden,W.J., Spear,E.D., Barlowe,C. and Ng,D.T.W. (2001) Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol., 155, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. and Chang,A. (1999) Eps1, a novel PDI-related protein involved in ER quality control in yeast. EMBO J., 18, 5972–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. and Chang,A. (2002) Sphingoid base synthesis is required for oligomerization and cell surface stability of the yeast plasma membrane ATPase, Pma1. Proc. Natl Acad. Sci. USA, 99, 12853–12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Meyer,H.H. and Rapoport,T.A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature, 414, 652–656. [DOI] [PubMed] [Google Scholar]