

Abstract
Agonist-promoted desensitization of the heterodimeric metabotropic GABAB receptor was investigated. Whereas no desensitization was observed in HEK293 cells heterologously expressing the receptor, GABA and the synthetic agonist baclofen induced a robust desensitization in cerebellar granule cells endogenously expressing the receptor. Taking advantage of this cell-specific desensitization phenotype, we identified GRK4 as the kinase involved in the neuronal desensitization. Transfection of small interference RNA directed against GRK4 significantly reduced GRK4 levels in cerebellar granule cells and strongly inhibited the agonist-promoted desensitization. Reciprocally, transfection of GRK4 in HEK293 cells restored agonist-promoted desensitization, confirming that this kinase is sufficient to support desensitization. Surprisingly, this desensitization occurred in the absence of ligand-induced receptor phosphorylation and could be promoted by GRK4 mutants deleted of their kinase domain. Taken together, these results suggest that GRK4 plays a central role in the agonist-promoted desensitization of GABAB receptor and that it does so through an atypical mechanism that challenges the generally accepted model linking the kinase activity of GRKs to their role in receptor desensitization.
Keywords: desensitization/GABAB receptor/GRK4
Introduction
G-protein-coupled receptors (GPCRs) play key roles in hormonal and neuronal regulation. Upon stimulation, they engage various signaling pathways that initiate cellular responses. Simultaneously, processes that regulate the signaling efficacy of the receptors themselves come into place and contribute to phenomena such as tolerance and tachyphylaxis. One of these processes, known as homologous desensitization, is characterized by the fact that the response of a given receptor wanes over time despite the continuous presence of the stimulus. For many GPCRs, the desensitization process appears to be primarily mediated by two protein families: the G-protein receptor kinases (GRKs) and arrestins (Ferguson et al., 1996). According to the generally accepted model, GRKs specifically bind to the agonist-occupied receptor and catalyze its phosphorylation. This, in turn, increases the affinity of the receptor for arrestin that binds the receptor, precluding receptor/G-protein interaction. In many instances, arrestin binding also leads to the removal of the receptor from the plasma membrane via clathrin-mediated endocytosis (Goodman et al., 1997; Krupnick et al., 1997; Laporte et al., 1999, 2000).
The multigene family of GRKs consists of seven members, GRK1–7, which are classified into three subfamilies on the basis of their sequence homology (Pitcher et al., 1998a). GRK1 (rhodopsin kinase) and GRK7 belong to the first subfamily, GRK2 and -3 form the second (βARK subfamily), while GRK4, -5 and -6 compose the third subfamily (GRK4 subfamily). GRK2, -3, -5 and -6 are widely distributed in different cell types and tissues, where they have been proposed to mediate homologous desensitization of a variety of GPCRs. In contrast, GRK1, -4 and -7 have more restricted tissue distribution. GRK1 and -7 are restricted to retina, where they phosporylate opsins and contribute to desensitization to light in rods and cones, respectively (Hargrave and McDowell, 1992; Weiss et al., 2001). For its part, GRK4 has been shown to be highly expressed in testis and its mRNA has been detected by PCR in brain and kidney (Ambrose et al., 1992; Sallese et al., 1994, 1997; Virlon et al., 1998), suggesting effective receptor substrate selectivity for this kinase.
Although the role of GRKs has been abundantly studied for the desensitization of class 1 GPCRs (to which rhodopsin and the β2-adrenegic receptor belong), less is known about the desensitization processes of class 2 and 3 receptors. For class 3, GRK2 and -4 were recently shown to contribute to the desensitization of the metabotropic glutamate receptor type 1 (mGluR1) (Dhami et al., 2002; Iacovelli et al., 2003) but little is known concerning the molecular mechanisms involved in the desensitization of the metabotropic GABAB receptor (GBR). Although the acute effects of GBR activation are well known, only a few studies have addressed the question of its desensitization. In a recent study, Couve et al. (2002) suggested that the decline in GABA-evoked potassium currents occurring following sustained stimulation results from receptor desensitization. However, they found that GBR phosphorylation by PKA inhibits the desensitization, challenging the accepted view that phosphorylation is a universal negative modulator of GPCRs. Studying the desensitization of GBR may be of particular interest when considering its atypical structural organization. Indeed, GBR is one of only a few GPCRs that function as obligatory heterodimers (the other examples being the taste T1R1 and T1R3 receptors; Nelson et al., 2002). Whether such heterodimeric receptors follow the classical desensitization paradigm thus remains to be investigated. The importance of GBR in mediating slow synaptic inhibition in the central nervous system (Couve et al., 2000; Billinton et al., 2001) further justifies studying the mechanism regulating its signaling efficacy. Therefore, the present study was designed to investigate the occurrence of GBR homologous desensitization in a heterologous expression system, as well as in neurons endogenously expressing the receptor.
Results
Lack of GBR desensitization in HEK293 cells
In a first attempt to determine whether GBR is subject to classical homologous desensitization in HEK293 cells, we investigated the ability of this Gi/Go coupled receptor to activate G-proteins after a sustained agonist stimulation. To this aim, the GABA-induced GTPγS binding was assessed in HEK293 cells transiently transfected with the two subunits, GBR1b and GBR2, required to form a functional GBR. As can be seen in Figure 1A, a 60 min pre-stimulation with GABA did not affect agonist- promoted GTPγS binding. This contrasts with the robust desensitization observed in cells expressing the δ-opioid receptor (δOR), another Gi/Go coupled receptor used as a positive control, where a 60 min pre-stimulation with the selective agonist SNC80 led to a 47.0 ± 5.1% reduction in the maximal agonist-stimulated GTPγS binding (Figure 1B). These results therefore suggest that, unlike δOR, GBR does not undergo rapid desensitization in HEK293 cells. As will be discussed below, the resistance to desensitization was also observed when baclofen, another GBR agonist, was used (see Figure 5A).

Fig. 1. Lack of agonist induced desensitization of GBR in HEK293 cells. (A and B) Agonist-stimulated [35S]GTPγS binding was measured in membranes derived from HEK293 cells expressing Myc-GBR1b/HA-GBR2 (A) or Myc-δOR (B) pre-stimulated (PS; triangles) or not (CTL; squares) for 60 min with the specific agonist [1 mM GABA (A) or 10 µM SNC80 (B)]. Data were analyzed by non-linear regression (GraphPad Prism) and are presented as the mean ± SEM of at least three independent experiments performed in triplicate. *P < 0.05 between the asymptotes (maximal response) of the curves. (C) The phosphorylation state of Myc-GBR1b and HA-GBR2 was studied following metabolic labeling with [32P]Pi. Cells transfected with pcDNA3 alone (mock) or Myc-GBR1b/HA-GBR2 were incubated (+) or not (–) with 1 mM GABA for 60 min and the receptor purified by immunoprecipitation (IP) using mouse anti-Myc (9E10) or anti-HA (12CA5) antibodies. Immunocomplexes were analyzed by SDS–PAGE and autoradiography. The identity of the bands was confirmed by western blot analysis (WB) on the same gels using rabbit anti-Myc (A14) or anti-HA (Y11) antibodies. The autoradiograms shown are representative of three independent experiments. Arrows indicate the phosphorylated forms of Myc-GBR1b and HA-GBR2. (D) Recruitment of β-arrestin in living cells. HEK293 cells were transiently co-transfected with Myc-V2R-Rluc or Myc-GBR1b-Rluc/HA-GBR2-Rluc in combination with β-arrestin-GFP10. For each pair considered, the quantities of DNA used for Myc-V2R-Rluc or Myc-GBR1b-Rluc/HA-GBR2-Rluc were selected to yield equivalent luminescent signals (370–450 nm), whereas that for β-arrestin-GFP10 was selected to obtain the maximum BRET levels. Cells were treated (agonist) or not (CTL) with the appropriate selective agonists (1 µM Arg-vasopressin and 1 mM GABA). Following 30 min incubation, the energy transfer reaction was initiated by adding DeepBlueC to each well and BRET assessed in a BRETCount microplate reader. The results represent the mean ± SEM of three to six independent experiments performed in triplicate. *P < 0.05.

Fig. 5. GRK4 promotes phosphorylation-independent desensitization of GBR in HEK293. (A and B) Baclofen-stimulated [35S]GTPγS binding was measured in membranes of HEK293 cells transfected with Myc-GBR1b/HA-GBR2 (A) or in combination with GRK4 (B) pre- stimulated (PS; triangles) or not (CTL; squares) for 60 min with the specific agonist (1 mM baclofen). Data represent the mean ± SEM of three independent experiments performed in triplicate. *P < 0.05. (C) Receptor phosphorylation was assessed following [32P]Pi metabolic labeling of HEK293 cells co-transfected with Myc-GBR1b/HA-GBR2 in the presence or absence of GRK4. Cells were either stimulated or not with 1 mM GABA for the indicated times. GBR1 and GBR2 were then purified by immunoprecipitation and resolved on SDS–PAGE as in Figure 1C. GBR phosphorylation is expressed as a percentage of the phosphorylation level observed in the absence of agonist and was normalized as a function of the total amount of purified receptor detected by western blot analysis carried out on the same blots. Data shown are the mean ± SEM of three independent experiments. (D) Recruitment of β-arrestin in living cells. HEK293 cells were transiently transfected with the indicated combination of plasmids. BRET experiments were then carried out as in Figure 1D. The results represent the mean ± SEM of three to six independent experiments performed in triplicate. (E) ELISA was performed to quantify cell surface receptor expression following agonist stimulation as in Figure 2C. *P < 0.05.
To further investigate this resistance to agonist- promoted desensitization, each of the steps involved in classical desensitization were assessed. First, the phosphorylation state of the receptor was determined following metabolic labeling experiments with [32P]Pi carried out in HEK293 cells co-expressing Myc-tagged GBR1b and HA-tagged GBR2. For this purpose, receptors were immunopurified from cells stimulated or not with GABA for 60 min. Both receptors were found to be phosphorylated under basal condition but, surprisingly, agonist stimulation did not promote any additional phosphorylation (Figure 1C).
Given the important role that receptor phosphorylation plays in promoting β-arrestin/receptor interaction, we then monitored β-arrestin recruitment by GBR using bioluminescence resonance energy transfer (BRET). This technique is a proximity assay based on the non-radiative transfer of energy between a bioluminescent donor (Renilla Luciferase, Rluc) and a fluorescent acceptor [green fluorescent protein (GFP)] that allows real-time monitoring of protein–protein interactions in living cells (Xu et al., 1999; Angers et al., 2000). β-arrestin–GFP and Rluc-tagged receptors (GBR1b-Rluc, GBR2-Rluc) were used for the assay. The Rluc-tagged V2 vasopressin receptor (V2R-Rluc), known to efficiently recruit β-arrestin, was used as a positive control. Whereas stimulation of V2R-Rluc with vasopressin promoted a significant increase in the BRET signal, reflecting the recruitment of GFP–β-arrestin to the receptor, no such BRET signal was observed upon GBR stimulation (Figure 1D), suggesting a lack of GFP–β-arrestin recruitment by the GBR1b-Rluc and GBR2-Rluc. Interestingly, although both receptor subtypes were phosphorylated at the basal level, no constitutive β-arrestin recruitment could be detected by BRET.
Consistent with the lack of agonist-promoted phosphorylation and β-arrestin recruitment, sustained stimulation did not induce GBR internalization. Indeed, immunocytochemical labeling of Myc-GBR1 and HA-GBR2 revealed no qualitative change in cell surface expression of this receptor following GABA stimulation for 5, 30 or 60 min. In contrast, β2-adrenergic receptor (HA-β2AR), Myc-δOR and Myc-V2R, all showed punctuate labeling corresponding to time-dependent internalization following selective agonist stimulation (Figure 2A). The lack of GBR endocytosis was further confirmed in cells co-expressing Myc-GBR1b, GBR2 and HA-β2AR, where HA-β2AR but not Myc-GBR1b was internalized after the addition of both isoproterenol and GABA (Figure 2B). A more quantitative assessment of GBR cell surface expression was carried out using an enzyme-linked immunosorbent assay (ELISA) in non-permeabilized HEK293 cells expressing Myc-GBR1b and HA-GBR2. As shown in Figure 2C, no significant change in receptor cell surface expression was observed even after 60 min of GABA stimulation, whereas the Myc-V2R cell surface expression decreased as early as 5 min and reached a minimum after 30 min of agonist stimulation.

Fig. 2. Lack of agonist-induced internalization of GBR in HEK293 cells. (A and B) Internalization of HA-β2AR, Myc-V2R, Myc-δOR or Myc-GBR1b/GBR2 expressed individually (A) and of Myc-GBR1b/GBR2 and HA-β2AR expressed simultaneously (B) was assessed by immunofluorescence microscopy. Intact cells were incubated with anti-Myc (9E10) or anti-HA (Y11) antibodies before stimulation with their specific agonists for the indicated time (β2AR: 1 µM isoproterenol; V2R: 1 µM Arg-vasopressin; δOR: 10 µM SNC80; and GBR: 1 mM GABA). Immunoreactivity was revealed with the appropriate Alexa488- or Alexa594-conjugated secondary antibodies. Characters in bold indicate the visualized receptors. Pictures shown are representative of five independent experiments. (C) ELISA was performed to quantify cell surface receptor expression following agonist stimulation. Results are expressed as a percentage of N-terminally tagged Myc-GBR1b/HA-GBR2 and Myc-V2R remaining at the cell surface following the indicated time of agonist stimulation, 0 min being taken as 100%. Data represent the mean ± SEM of three independent experiments. *P < 0.05.
GRK4-induced desensitization of GBR in cerebellar granule cells
To determine whether the lack of GBR desensitization is an intrinsic property of the GBR or rather reflects the absence, in the HEK293 cells, of proteins specifically involved in its desensitization, the ability of GBR to undergo desensitization was assessed in cerebellar granule cells. The endogenous expression of GBR in these cells (Ige et al., 2000) was confirmed by anti-GBR2 immunolabeling experiments revealing a somatic and neuritic punctuate distribution pattern (Figure 3A). Since these cells also express the ionotropic GABA receptor, desensitization was assessed using the metabotropic selective ligand baclofen instead of GABA to avoid confounding effects. Interestingly, when cerebellar granule cells were pre-stimulated with 1 mM baclofen for 60 min, the GTPγS binding induced by subsequent baclofen application was largely reduced, such that a 67.2 ± 4.8% desensitization was measured (Figure 3B). These results therefore show that GBR is not intrinsically resistant to desensitization but that a specific factor(s) present in the cerebellar granule neurons is lacking in HEK293 cells.
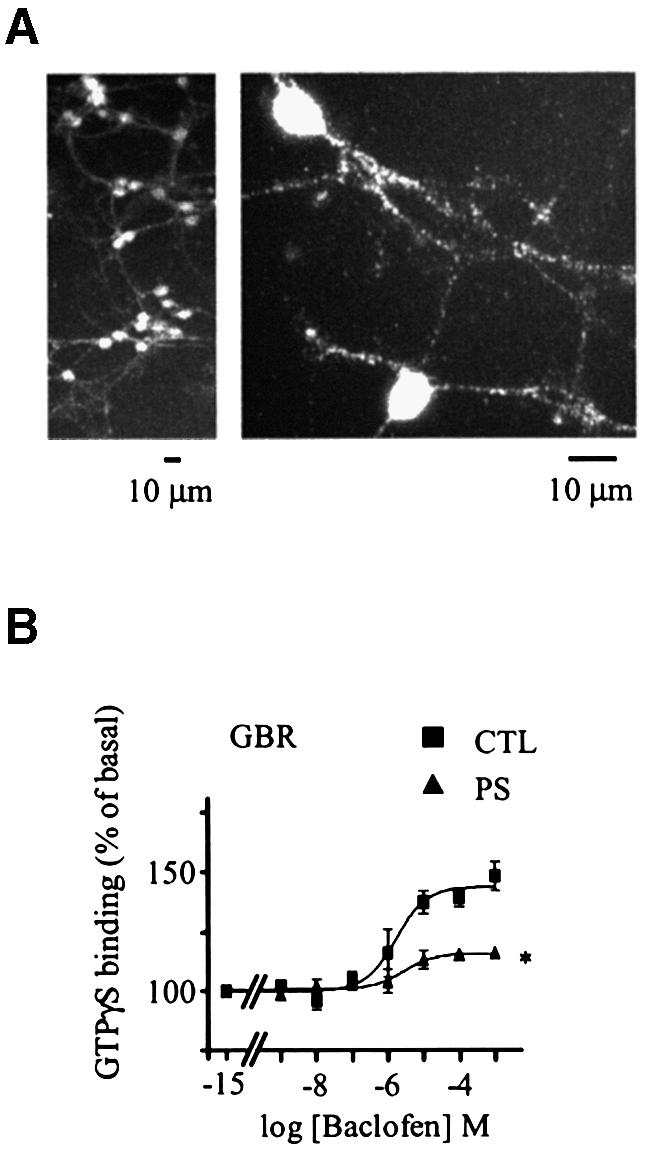
Fig. 3. Desensitization of GBR in cerebellar granule cell. (A) Endogenously expressed GBRs were detected in cerebellar granule cells (GCG) by immunofluorescence microscopy. Permeabilized cells were immunolabelled using a polyclonal antibody raised against native GBR2. (B) Baclofen-stimulated [35S]GTPγS binding was measured in membranes derived from CGC, pre-stimulated (PS; triangles) or not (CTL; squares) for 60 min with the selective agonist (1 mM baclofen). Data represent the mean ± SEM of three independent experiments performed in triplicate. *P < 0.05.
Given the predominant role played by GRKs in the desensitization of many GPCR, the fact that one subtype, GRK4, was lacking in HEK293 cells but expressed in cerebellar granule cells (Figure 4A) made it a likely candidate involved in GBR desensitization. To test directly the potential role of GRK4 in GBR desensitization, small interference RNA (siRNA) targeted against GRK4 mRNA (GRK4-siRNA) was used. As shown in Figure 4B, treatment with GRK4-siRNA led to a concentration-dependent decrease in GRK4 protein expression level that reached 30% of the control value at the largest siRNA concentration used. This decrease in GRK4 protein expression level was accompanied by a blunting of the baclofen-induced GBR desensitization. Indeed, the 65.6 ± 6.0% desensitization observed in untreated cells was reduced to 32.1 ± 4.6% in cerebellar granule cells transfected with the GRK4-siRNA (Figure 4C and D). GRK4 thus appears to play a crucial role in promoting GBR homologous desensitization in cerebellar granule cells.

Fig. 4. The GRK4 protein is required for GBR desensitization in cerebellar granule cells. (A) Total GRK4 immunoreactivity was assessed in total lysate from HEK293 cells (transfected with Myc-GBR1b/HA-GBR2 alone or with GRK4) and in native cerebellar granule cells (CGC). (B) Total GBR2 and GRK4 immunoreactivity detected in total lysate from CGC transfected or not with the indicated amount of GRK4-siRNA. In (A) and (B), proteins were detected following SDS–PAGE and immunoblotting using rabbit anti-GRK4 (H70) or rabbit anti-GBR2 antibodies. The western blots shown are representative of three independent experiments. (C) Baclofen-stimulated [35S]GTPγS binding was measured in membranes of CGC transfected or not with 100 nM GRK4-siRNA and pre-stimulated (PS) or not (CTL) for 60 min with 1 mM baclofen. Data represent the mean ± SEM of three independent experiments performed in triplicate. (D) The desensitization of the responses measured in (C) was expressed as the decrease in baclofen-stimulated response following pre-stimulation. The data are expressed as percentage of the control response. *P < 0.05.
To further confirm the role of GRK4 in GBR homologous desensitization, HEK293 cells were transfected with GRK4 cDNA and the GBR desensitization assessed. Whereas no desensitization was observed in the HEK293 cells following a 60 min baclofen pre-treatment (Figure 5A), the agonist promoted a 48.1 ± 4.8% desensitization in cells co-expressing GBR and GRK4 (Figure 5B), indicating that GRK4 is sufficient to support GBR homologous desensitization. In contrast, co-transfection of the related GRK6 did not favor baclofen-promoted desensitization, thus supporting the selectivity of the effects observed with GRK4 (data not shown).
Phosphorylation-independent GBR desensitization by GRK4
Since the above results demonstrated the role of GRK4 in GBR desensitization, we next investigated whether receptor phosphorylation accompanied desensitization. Metabolic labeling experiments with [32P]Pi were carried out in cells co-expressing Myc-GBR1b, HA-GBR2 and GRK4. Unexpectedly, even in the presence of GRK4, no agonist-induced GBR phosphorylation was observed (Figure 5C). In fact, the overexpression of GRK4 did not affect the phosphorylation state of GBR1 or GBR2, whether in the presence or absence of agonist stimulation. Consistent with this result, GRK4 expression had no effect on β-arrestin recruitment assessed by BRET (Figure 5D) and did not promote GBR internalization (Figure 5E) upon GABA stimulation.
The apparent independence of the GBR desensitization from its phosphorylation state challenged the generally accepted model linking the kinase activity of GRKs to their role in receptor desensitization. To formally test the role of GRK4 kinase activity in the GBR desensitization process, we took advantage of the conserved topology characterizing the GRK family. These proteins are subdivided into three functional domains: an N-terminal domain mediating receptor interaction and containing an RGS homology domain, a central catalytic domain and a highly variable C-terminal domain required for membrane targeting of the kinases (Premont et al., 1995; Figure 6A). Thus, we tested the ability of a GRK4 construct missing its central catalytic domain (Δ173–445; GRK4-delKD) to support agonist-promoted desensitization (Figure 6A). As shown in Figure 6B, this mutant form of GRK4 was sufficient to promote a loss of GTPγS binding upon sustained stimulation with baclofen, indicating that the catalytic domain of GRK4 is not necessary to promote GBR homologous desensitizaton. The GRK4-delKD was also found to interact with GBR, since both wild-type (wt) and GRK4-delKD could be co-immunoprecipitated with GBR1b (not shown) and GBR2 (Figure 6B) in cells co-expressing the two receptor subtypes. Co-immunoprecipitation between GBR2 and both GRK4 and GRK4-delKD was observed even in the absence of agonist stimulation, indicating a level of constitutive interaction. Furthermore, agonist stimulation increased the interaction between the receptor and GRK4-wt or GRK-delKD by 27.1 ± 2.2 and 30.0 ± 3.6%, respectively.

Fig. 6. GRK4 interacts with and promotes the desensitization of GBR independently of its kinase domain. (A) Topological organization of GRK4 and GRK4-delKD. (B and C) Baclofen-stimulated [35S]GTPγS binding was measured in membranes of HEK293 cells transfected with Myc-GBR1b/HA-GBR2 and GRK4 (B) or GRK4-delKD (C) pre- stimulated (PS; triangles) or not (CTL; squares) for 60 min with the specific agonist (1 mM baclofen). Data represent the mean ± SEM of three experiments performed in triplicate. *P < 0.05. (D) Left panel, co-immunoprecipitation of GRK4 and GRK4-delKD with HA-GBR2 was carried out in HEK293 cell lysates transfected with the indicated plasmids. Immunoprecipitation (IP) using anti-HA (12CA5) antibody was performed on total cell extracts pre-treated or not with 1 mM GABA for 60 min. The immunocomplexes were then analyzed by western blot analysis using antibody (H70) raised against the N-terminal part of GRK4 (common to both GRK4 constructs). Right panel: following HA-GBR2 IP, GRK4 immunoreactivity was quantified by laser densitometry and the data analyzed using Quantity One (Bio-Rad). Arrows indicate the bands corresponding to GRK4 and GRK4-delKD. Data shown are the mean ± SEM of three independent experiments. *P < 0.05.
Discussion
In the present study we report that GBR heterologously expressed in HEK293 cells does not undergo rapid desensitization and supports sustained responses upon agonist stimulation. However, this is not an intrinsic property of the receptor, since a robust desensitization is observed in cerebellar granule cells endogenously expressing GBR. Taking advantage of this cell-type-specific phenotype, we identified GRK4 as a regulatory protein required to promote GBR desensitization. Surprisingly, this GRK4-dependent desensitization occurs in the absence of an increased receptor phosphorylation and does not require the catalytic activity of the kinase. This obviously contrasts with the desensitization paradigm accepted for a large number of receptors and suggests that, in addition to its kinase activity, GRK4 possesses additional regulatory properties.
GBR can thus be added to a short list of GPCRs, including the luteinizing hormone/human chorionic gonadotropin receptor (Munshi et al., 2001), the dopaminergic D1 receptors (Watanabe et al., 2002) and mGluR1 (Sallese et al., 2000b; Iacovelli et al., 2003), found to be regulated by GRK4. It should, however, be noted that the suggested role of GRK4 in the desensitization of mGluR1 in HEK293 and cerebellar Purkinje cells (Sallese et al., 2000b) was questioned in a more recent study where overexpression of GRK2 and -5 but not GRK4 and -6 promoted mGluR1 desensitization in HEK293 cells. In any case, although the full contingent of receptors regulated by GRK4 is far from being completely established, the restricted pattern of expression of the kinase may suggest that it will encompass a smaller subset of receptors than those regulated by the ubiquitously expressed GRK2. To date, northern blot analysis has revealed high GRK4 mRNA expression only in testis, where the presence of the protein was also confirmed (Premont et al., 1996; Sallese et al., 1997). However, RT–PCR revealed mRNA signals in brain (Sallese et al., 1994; Weiss et al., 2001) and kidney (Sallese et al., 1994; Virlon et al., 1998). In situ hybridization has also suggested GRK4 expression in rat cerebellum, where it was found in Purkinje but not granule cells (Sallese et al., 2000b). This lack of mRNA expression in rat cerebellar granule cells contrasts with our immunodetection of the GRK4 protein in the same cells in mice. Whether this reflects a true species difference in the tissue distribution of GRK4 remains to be investigated.
Although our results clearly demonstrate that GRK4 is crucial in cerebellar neurons and sufficient in HEK293 cells to promote GBR desensitization, they point to a non-canonical mechanism of action for the kinase. Indeed, a GRK4 mutant lacking the kinase catalytic domain (Δ173–445; GRK4-delKD; Figure 6C) was found to support agonist-promoted desensitization. Similar results were also obtained with a truncated GRK4 construct lacking the catalytic and the entire C-terminal domains (GRK4 1–172trunc), demonstrating that the N-terminal domain is sufficient to promote GBR desensitization (data not shown). Combined with the lack of agonist-promoted GBR phosphorylation even in the presence of the full-length GRK4 (Figure 5C), these results suggest that GBR desensitization occurs in a phosphorylation-independent manner not involving β-arrestin recruitment (Figure 5D) or receptor internalization (Figure 5E). Since the absence of the catalytic domain did not inhibit the agonist-promoted recruitment of GRK4 to the receptor (Figure 6D), one could hypothesize that the physical interaction between these two proteins is sufficient to hinder receptor coupling to its downstream effectors. Consistent with this notion is the observation that the interaction between GBR and GRK4 is quite stable, as illustrated by the significant co-immunoprecipitation of GRK4 with GBR2 even in the absence of receptor activation. The relatively high affinity of GRK2 for specific receptors has previously been used to explain its phosphorylation-independent influence on signaling efficacy (Diviani et al., 1997; Freedman et al., 1997). Indeed, the ability of the overexpressed GRK2-K220R mutant, which lacks kinase activity, to inhibit phospholipase C stimulation by endothelin-1 and α1b-adrenergic receptors was correlated with the ability of these receptors to be co-immunoprecipitated with the kinase. The extent of co-immunoprecipitation between GBR and GRK4 observed in the present study could also be taken as evidence for a relatively high affinity between these two proteins, consistent with the contribution of steric hindrance to the desensitization process. The fact that GRK4 is constitutively associated to the plasma membrane, most likely through its palmitate moieties (Premont et al., 1996), may increase its apparent affinity for the receptor and contribute to this non-canonical desensitization process.
However, a role as a scaffolding organizer that facilitates interactions with additional regulatory proteins or sequesters effector protein away from the receptor cannot be excluded. Indeed, GRKs have been proposed to interact with many proteins able to influence signaling efficacy. These include: G protein α (Carman et al., 1999b; Sallese et al., 2000a; Usui et al., 2000) and βγ subunits (Pitcher et al., 1992; Carman et al., 2000), clathrin (Shiina et al., 2001), the GRK-interacting protein GIT1 (Premont et al., 1998), caveolin (Carman et al., 1999a), phosphoinositide 3-kinase α and γ (Naga Prasad et al., 2001), and cytoskeletal (Pitcher et al., 1998b) and calcium binding (Iacovelli et al., 1999) proteins.
The N-terminal portion of GRK2 originally proposed as the interaction site for GPCRs (Premont et al., 1995) has more recently been shown to possess an RGS homology domain that interacts with and modestly catalyzes the GTPase activity (GAPing) of Gαq (Carman et al., 1999b). This domain of GRK2 was also shown to inhibit receptor-stimulated phospholipase C activity, suggesting that GRK2 can contribute to signal termination through a direct interaction with Gαq. The relatively modest GAPing activity compared with the dramatic inhibitory effect on the phospholipase C led the authors to suggest that the GRK2 RGS domain acts mainly by sequestering the Gαq away from the effector. Such a mechanism was also proposed to explain the ability of the kinase-dead GRK2-K220R/W mutants to inhibit receptor-activated phospholipase C for several Gq-coupled receptors (Carman et al., 1999b; Usui et al., 2000; Dhami et al., 2002; Iacovelli et al., 2003). Although the phosphorylation-independent inhibition of Gq-mediated signaling has been interpreted as a possible mechanism contributing to GRK2-mediated desensitization, none of these studies directly tested its role in a classical agonist-promoted desensitization paradigm. This was done in only one study, where the expression of GRK2-K220R was shown to potentiate the agonist-promoted desensitization of the PTH-stimulated GTPase activity as efficiently as the wild-type GRK2 (Dicker et al., 1999).
Given the overall topological conservation between the GRKs, it could be hypothesized that the N-terminal domain of GRK4 could contribute to GBR desensitization in a similar manner. Indeed, GRK4 binding to Gαi/o could sequester the G protein subunit away from the receptor, thus preventing functional re-coupling that would be detected as a decrease in subsequent GABA-stimulated GTPγS binding. However, whether sequestration of Gαi/o, sustained binding to the receptor or both truly contribute to the GRK4-mediated desensitization of GBR remains to be investigated.
Independently of the detailed mechanism involved, the present study clearly shows that phosphorylation- independent action of GRKs is not limited to GRK2. The finding that GRK4 can also promote desensitization independently of receptor phosphorylation could suggest that such action is a general characteristic of this class of regulatory proteins. It should also be pointed out that this is the first demonstration of a phosphorylation-independent desensitization for a non-Gq-coupled receptor, since GBR is preferentially coupled to Gi/o. Whether GRK4 will be found to regulate other GPCRs in a kinase-independent manner remains to be investigated. Additional studies will also be required to determine the functional implications of the existence of two GRK-mediated desensitization processes, only one involving receptor phosphorylation.
Materials and methods
HEK293 cell culture and transfection
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and streptomycin, 2 mM l-glutamine (all from Wisent). For transfection experiments, cells were seeded at a density of 2 × 106 cells per 100 mm dish and cultured for 24 h. Transient transfections were then performed using the calcium phosphate precipitation method (Mellon et al., 1981). Twenty-four hours after transfection, DMEM was renewed and cells cultured for an additional 24 h.
Cerebellar granule cell culture and siRNA transfection
Primary cultures of cerebellar cells were prepared as described previously (Van Vliet et al., 1989). Briefly, 1-week-old newborn mice were decapitated and cerebellum dissected. The tissue was then gently triturated using fire-polished Pasteur pipettes and the homogenate centrifuged at 30 g. The pellet was resuspended and plated in tissue culture dishes previously coated with poly-d-lysine. Cells were maintained in a neurobasal medium (B27; Gibco-BRL) supplemented with 25 µM glutamate, 500 µM glutamine, 10% decomplemented fetal calf serum (FCS) and 25 mM KCl to improve neuronal survival. One-week-old cultures contained 125 × 103 cells/cm2.
The GRK4-siRNA was designed, as recommended (Elbashir et al., 2001b), with 5′ phosphate, 3′ hydroxyl and two bases overhang on each strand. siRNA was chemically synthesized by Xeragon. The gene-specific sequence TGGAGAGAGCTCCTGAAGT [from bases 85 to 103 of the Mus musculus GRK4 mRNA (DDBJ/EMBL/GenBank accession No. AF040745) sequence] was specifically selected not to target other potentially cross-reactive GRKs and was absent from the closely related GRK5 and GRK6. Annealing for duplex siRNA formation was performed as described previously (Elbashir et al., 2001a). Transfections of siRNA (50–200 nM for western blot analyses and 100 nM for GTPγS binding experiments) in cerebellar granule cells were carried out with Transfast transfection reagent (Promega) according to Ango et al. (1999). Cells were analyzed 1 week following transfection.
Plasmids
pcDNA3.1-Myc-GBR1b and pcDNA3.1-HA-GBR2 plasmids were a generous gift from GlaxoSmithKline. Briefly, the entire coding region of GBR1b or GBR2 was subcloned in the Myc-containing plasmid (pSigMyc) or the HA-containing plasmid (pCIN6), respectively, in order to add the desired tag at the N-terminus of each receptor. The resulting sequence was subsequently transferred into the pcDNA3.1 plasmid (Invitrogen). Additional receptor plasmids used include pcDNA3.1-HA-β2AR (Lavoie et al., 2002), Myc-V2R (Morello et al., 2000) and Myc-δOR (Petaja-Repo et al., 2002).
The human pcDNA3-GRK4 and GRK6 plasmids were generously provided by Dr Antonio De Blasi (Istituto Neurologico Mediterraneo Neuromed, Pozzilli, Italy). The GRK4 mutant (GRK4-delKD) was constructed using a PCR strategy that removed the kinase domain of the protein (amimo acids 173–445).
For BRET experiments, GBRs tagged at their C-teminus with either Rluc or GFP10 were used. The coding sequences of Rluc or GFP10 were added in-frame to the 3′ end coding sequence of Myc-GBR1b, HA-GBR2 and Myc-V2R within their respective pcDNA3.1 plasmids. For β-arrestin2, the GFP10 was fused at the C-terminus (pcDNA3.1-β-arrestin-GFP10), while the Rluc was added in-frame to the N-terminus (pcDNA3.1-Rluc-β-arrestin) of the rat arrestin2.
[35S]GTPγS binding assay
Transiently transfected HEK293 cells or cerebellar granule neurons, pre-stimulated or not with the appropriate agonist for 60 min, were resuspended and lysed under hypotonic conditions (25 mM Tris–HCl pH 7.4, 5 mM MgCl2, 2 mM EDTA, 5 µg/ml leupeptin, 5 µg/ml soybean trypsin inhibitor and 10 µg/ml benzamidine) and homogenized with a polytron homogenizer (Ultra-Turrax; Janke and Kunkel) for 10 s. Homogenates were centrifuged at 500 g for 5 min at 4°C and the resulting supernatant fraction was centrifuged at 25 000 g for 20 min at 4°C. The membrane pellets were resuspended in assay buffer [50 mM HEPES pH 7.4, 200 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol (DTT), 15 µM GDP and 0.5% bovine serum albumin (BSA)]. Membranes (20 µg of protein per assay) were incubated in 300 pM [35S]GTPγS (1250 Ci/mmol) in the presence of various agonist concentrations for 60 min at 25°C. Binding was terminated by rapid filtration over GF/B filters using a cell harvester. Filters were washed three times in 50 mM Tris pH 7.4 containing 50 mM NaCl and 5 mM MgCl2, and counted.
Whole-cell phosphorylation
Following incubation for 60 min in phosphate-free medium containing 1% FCS, cells expressing Myc-GBR1b/HA-GBR2 were incubated for 2 h in phosphate-free medium containing [32P]Pi (1.7 mCi/106 cells) and treated or not with 1 mM GABA for 60 min. They were then solubilized in RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 2.5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 1 mM DTT, 1% SDS, 5 µg/ml leupeptine, 5 µg/ml soybean trypsin inhibitor and 10 µg/ml benzamidine) containing 0.2 mM sodium orthovanadate, 10 mM sodium fluoride, 10 mM sodium phosphate and centrifuged at 12 000 g for 15 min at 4°C. Immunoprecipitation was then performed as described below using monoclonal anti-Myc antibody (9E10) or monoclonal anti-HA antibody (12CA5). Proteins were resolved using 8% SDS–PAGE, transferred to nitrocellulose, and [32P] was detected by autoradiography using BioMax MR Kodak films (Perkin Elmer).
Immunoprecipitation
Cells grown in 100-mm Petri dishes were washed twice with phosphate-buffered saline (PBS), lysed for 20 min at 4°C in RIPA buffer (see above) and centrifuged at 12 000 g for 15 min at 4°C. Immunoprecipitation was initiated by adding mouse anti-Myc (clone 9E10) or mouse anti-HA (12CA5) antibodies and protein G–Sepharose to the supernatant. Following an overnight incubation at 4°C, immunoreactive complexes were collected by centrifugation and washed four times with ice-cold RIPA buffer containing 350 mM NaCl. The final pellet was resuspended in sample buffer containing 60 mM Tris–HCl pH 6.8, 2% SDS, 4.5 M urea with 100 mM DTT, and western blot analyses peformed. For co-imunoprecipitation studies (Figure 6D), proteins were solubilized in a less stringent buffer consisting of 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.1% Triton and 1 mM DTT.
Western blot analysis
Protein samples were resolved on 8% PAGE (or 14% for Figure 6D), transferred to nitrocellulose and subjected to immunoblotting using rabbit A14 polyclonal anti-Myc antibodies, rabbit Y11 polyclonal anti-HA antibodies (1:5000; both from Santa Cruz), rabbit H70 polyclonal anti-GRK4 antibodies (1:500; Santa Cruz) or a rabbit polyclonal anti-GBR2 antibodies (1:5000; kindly provided by GlaxoSmithKline UK) (Ige et al., 2000). The nitrocellulose was then incubated with anti-rabbit/horseradish peroxidase conjugate (1:10 000; Amersham Biosciences) for 30 min and developed using the Renaissance chemiluminescence kit (Perkin Elmer). The selectivity of the H70 anti-GRK4 antibody was established by the lack of immunoreactivity in untransfected HEK293 cells and the unique band detected in cells transfected with the human GRK4 (Figure 4A). Its ability to recognize the mouse GRK4 in cerebellar granule cells was further confirmed by the observation that transfection of the GRK4-siRNA led to a >75% reduction in immunoreactivity detected by this antibody (Figure 4B).
BRET measurement
Forty-eight hours post-transfection, cells were washed twice with PBS, detached with PBS/EDTA and resuspended in PBS/0.1% glucose. Protein concentration was determined using the Bradford assay kit (Bio-Rad) and BSA as standard to control for cell number.
The β-arrestin recruitment assay was carried out as in Angers et al. (2000) using a slightly modified BRET technique (BRET2) described recently (Mercier et al., 2002). Briefly, cells expressing receptor–Rluc and β-arrestin–GFP constructs (Figure 1D) or receptor–GFP and Rluc–β-arrestin (Figure 5D) were distributed in 96-well microplates (white Costar plate with clear bottom) at a density of ∼100 000 cells/well. Cells were then treated or not with 1 mM GABA for 10 min before adding DeepBlueC coelenterazine at a final concentration of 5 µM. Readings were collected using a modified Top-count instrument (BRETCount; Packard Bioscience) that allows the sequential integration of the signals detected in the 370–450 and 500–530 nm windows using filters with the appropriate band pass (Chroma). The BRET signal is determined by calculating the ratio of the light emitted by the GFP (500–530 nm) over the light emitted by the Rluc (370–450 nm). The values were corrected by subtracting the background signal detected when the Rluc constructs were expressed alone.
Immunolabeling in permeabilized cells
Cerebellar granule cells were fixed with 3% paraformaldehyde in PBS for 15 min and permeabilized for 10 min with 0.25% Triton X-100 in blocking buffer (PBS containing 0.2% BSA). Cells were then incubated with a rabbit anti-GBR2 antibody (1:1000) in blocking buffer for 30 min, washed in blocking buffer and incubated with the anti-rabbit secondary antibody Alexa Fluor 594 (1:500; Molecular Probes) for 30 min in the dark. After extensive washing, the coverslips were mounted. Immunolabeling was viewed on a Zeiss fluorescence microscope using a Plan-Apo 63 × 1.40 NA oil immersion lens.
Internalization
HEK293 cells were transiently transfected with receptors tagged with the HA or Myc epitopes at their N-terminus. After washes with PBS, cells were incubated in DMEM/HEPES blocking buffer (20 mM HEPES pH 7.4, 0.2% BSA in DMEM) for 15 min. Rabbit anti-HA and/or mouse anti-Myc antibodies (1:100) were then added in the same buffer and incubated for 60 min at 4°C. Cells were then washed and treated with the appropriate agonist for different times at 37°C. The internalization process was stopped on ice and cells were fixed in 3% paraformaldehyde in PBS for 15 min. Cells were then prepared as before using the appropriate fluorophore-conjugated secondary antibodies for visualization of the receptors (Alexa Fluor 594 and Alexa Fluor 488 secondary antibodies; 1:500). Dual labeling was viewed on a Zeiss microscope using a Plan-Apo 63 × 1.40 NA oil immersion lens.
ELISA
Cells were transfected with receptors bearing a Myc or HA tag at their N-terminus and transferred to 12-well poly-d-lysine-coated plates 24 h after transfection. Forty-eight hours post-transfection, cells were treated with specific agonist or vehicle for different times at 37°C. After three washes with cold PBS, cells were fixed with 3% paraformaldehyde for 15 min and washed three times with PBS. Cells were blocked with PBS/1% BSA for 30 min, incubated with anti-Myc (9E10) or anti-HA (12CA5) antibodies (1:500) for 30 min followed by a 30 min incubation with anti-mouse/horseradish peroxidase conjugate (1:1000; Amersham Biosciences). All washes were performed three times with PBS/1% BSA. The substrate O-phenylenediamine dihydrochloride (Sigma) was then added according to the manufacturer’s instructions for 4–7 min, reaction stopped with 200 µl of 3 N HCl and the absorbance measured at 492 nm.
Statistical analysis
Statistical significance of the difference was determined using Student’s t-test analysis. A P value of 0.05 was used as the criterion for statistically significant differences.
Acknowledgments
Acknowledgements
The authors are grateful to Louise Cournoyer for her help in establishing the cerebellar granule cell cultures, Monique Vasseur for her assistance with immunofluorescence micrsocopy, and Drs Julia White, Alan Wise and Monique Lagacé for their critical reading of the manuscript. We also thank Neil Fraser and Piers Emson from GlaxoSmithKline for providing GBR1 and GBR2 constructs and the anti-GBR2 antibodies, as well as Dr Antonio De Blasi for providing us the GRK4 and GRK6 construct. This work was supported by a CIHR/Rx&D grant sponsored by GlaxoSmithKline. J.P. holds a fellowship from EMBO and M.B. is a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
References
- Ambrose C., James,M., Barnes,G., Lin,C., Bates,G., Altherr,M., Duyao,M., Groot,N., Church,D. and Wasmuth,J.J. (1992) A novel G protein-coupled receptor kinase gene cloned from 4p16.3. Hum. Mol. Genet., 1, 697–703. [DOI] [PubMed] [Google Scholar]
- Angers S., Salahpour,A., Joly,E., Hilairet,S., Chelsky,D., Dennis,M. and Bouvier,M. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl Acad. Sci. USA, 97, 3684–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F., Albani-Torregrossa,S., Joly,C., Robbe,D., Michel,J.M., Pin,J.P., Bockaert,J. and Fagni,L. (1999) A simple method to transfer plasmid DNA into neuronal primary cultures: functional expression of the mGlu5 receptor in cerebellar granule cells. Neuropharmacology, 38, 793–803. [DOI] [PubMed] [Google Scholar]
- Billinton A., Ige,A.O., Bolam,J.P., White,J.H., Marshall,F.H. and Emson,P.C. (2001) Advances in the molecular understanding of GABAB receptors. Trends Neurosci., 24, 277–282. [DOI] [PubMed] [Google Scholar]
- Carman C.V., Lisanti,M.P. and Benovic,J.L. (1999a) Regulation of G protein-coupled receptor kinases by caveolin. J. Biol. Chem., 274, 8858–8864. [DOI] [PubMed] [Google Scholar]
- Carman C.V., Parent,J.L., Day,P.W., Pronin,A.N., Sternweis,P.M., Wedegaertner,P.B., Gilman,A.G., Benovic,J.L. and Kozasa,T. (1999b) Selective regulation of Gα(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J. Biol. Chem., 274, 34483–34492. [DOI] [PubMed] [Google Scholar]
- Carman C.V., Barak,L.S., Chen,C., Liu-Chen,L.Y., Onorato,J.J., Kennedy,S.P., Caron,M.G. and Benovic,J.L. (2000) Mutational analysis of Gβγ and phospholipid interaction with G protein-coupled receptor kinase 2. J. Biol. Chem., 275, 10443–10452. [DOI] [PubMed] [Google Scholar]
- Couve A., Moss,S.J. and Pangalos,M.N. (2000) GABAB receptors: a new paradigm in G protein signaling. Mol. Cell. Neurosci., 16, 296–312. [DOI] [PubMed] [Google Scholar]
- Couve A., Thomas,P., Calver,A.R., Hirst,W.D., Pangalos,M.N., Walsh,F.S., Smart,T.G. and Moss,S.J. (2002) Cyclic AMP-dependent protein kinase phosphorylation facilitates GABAB receptor-effector coupling. Nat. Neurosci., 5, 415–424. [DOI] [PubMed] [Google Scholar]
- Dale L.B., Bhattacharya,M., Anborgh,P.H., Murdoch,B., Bhatia,M., Nakanishi,S. and Ferguson,S.G. (2000) G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J. Biol. Chem., 275, 38213–38220. [DOI] [PubMed] [Google Scholar]
- Dhami G.K., Anborgh,P.H., Dale,L.B., Sterne-Marr,R. and Ferguson,S.S. (2002) Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J. Biol. Chem., 277, 25266–25272. [DOI] [PubMed] [Google Scholar]
- Dicker F., Quitterer,U., Winstel,R., Honold,K. and Lohse,M.J. (1999) Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc. Natl Acad. Sci. USA, 96, 5476–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D., Lattion,A.L. and Cotecchia,S. (1997) Characterization of the phosphorylation sites involved in G protein-coupled receptor kinase- and protein kinase C-mediated desensitization of the α1B-adrenergic receptor. J. Biol. Chem., 272, 28712–28719. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001a) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Martinez,J., Patkaniowska,A., Lendeckel,W. and Tuschl,T. (2001b) Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J., 20, 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S.S., Barak,L.S., Zhang,J. and Caron,M.G. (1996) G-protein-coupled receptor regulation: role of G-protein-coupled receptor kinases and arrestins. Can. J. Physiol. Pharmacol., 74, 1095–1110. [DOI] [PubMed] [Google Scholar]
- Freedman N.J., Ament,A.S., Oppermann,M., Stoffel,R.H., Exum,S.T. and Lefkowitz,R.J. (1997) Phosphorylation and desensitization of human endothelin A and B receptors. Evidence for G protein-coupled receptor kinase specificity. J. Biol. Chem., 272, 17734–17743. [DOI] [PubMed] [Google Scholar]
- Goodman O.B. Jr, Krupnick,J.G., Gurevich,V.V., Benovic,J.L. and Keen,J.H. (1997) Arrestin/clathrin interaction. Localization of the arrestin binding locus to the clathrin terminal domain. J. Biol. Chem., 272, 15017–15022. [DOI] [PubMed] [Google Scholar]
- Hargrave P.A. and McDowell,J.H. (1992) Rhodopsin and phototrans duction: a model system for G protein-linked receptors. FASEB J., 6, 2323–2331. [DOI] [PubMed] [Google Scholar]
- Iacovelli L., Sallese,M., Mariggio,S. and De Blasi,A. (1999) Regulation of G-protein-coupled receptor kinase subtypes by calcium sensor proteins. FASEB J., 13, 1–8. [DOI] [PubMed] [Google Scholar]
- Iacovelli L. et al. (2003) Role of G protein-coupled receptor kinase 4 and β-arrestin 1 in agonist-stimulated metabotropic glutamate receptor 1 internalisation and activation of mitogen-activated protein kinases. J. Biol. Chem., 278, 12433–12442. [DOI] [PubMed] [Google Scholar]
- Ige A.O., Bolam,J.P., Billinton,A., White,J.H., Marshall,F.H. and Emson,P.C. (2000) Cellular and sub-cellular localisation of GABA(B1) and GABA(B2) receptor proteins in the rat cerebellum. Mol. Brain Res., 83, 72–80. [DOI] [PubMed] [Google Scholar]
- Krupnick J.G., Goodman,O.B.,Jr, Keen,J.H. and Benovic,J.L. (1997) Arrestin/clathrin interaction. Localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J. Biol. Chem., 272, 15011–15016. [DOI] [PubMed] [Google Scholar]
- Laporte S.A., Oakley,R.H., Zhang,J., Holt,J.A., Ferguson,S.S., Caron,M.G. and Barak,L.S. (1999) The β2-adrenergic receptor/ β-arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl Acad. Sci. USA, 96, 3712–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte S.A., Oakley,R.H., Holt,J.A., Barak,L.S. and Caron,M.G. (2000) The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated pits. J. Biol. Chem., 275, 23120–23126. [DOI] [PubMed] [Google Scholar]
- Lavoie C. et al. (2002) β1/β2-adrenergic receptor heterodimerization regulates β2-adrenergic receptor internalization and ERK signaling efficacy. J. Biol. Chem., 277, 35402–35410. [DOI] [PubMed] [Google Scholar]
- Mellon P., Parker,V., Gluzman,Y. and Maniatis,T. (1981) Identification of DNA sequences required for transcription of the human α1-globin gene in a new SV40 host-vector system. Cell, 27, 279–288. [DOI] [PubMed] [Google Scholar]
- Mercier J.F., Salahpour,A., Angers,S., Breit,A. and Bouvier,M. (2002) Quantitative assessment of β1- and β2-adrenergic receptor homo- and hetero-dimerization by bioluminescence resonance energy transfer. J. Biol. Chem., 277, 44925–44931. [DOI] [PubMed] [Google Scholar]
- Morello J.P. et al. (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest., 105, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi U.M., Peegel,H. and Menon,K.M. (2001) Palmitoylation of the luteinizing hormone/human chorionic gonadotropin receptor regulates receptor interaction with the arrestin-mediated internalization pathway. Eur. J. Biochem., 268, 1631–1639. [DOI] [PubMed] [Google Scholar]
- Naga Prasad S.V., Barak,L.S., Rapacciuolo,A., Caron,M.G. and Rockman,H.A. (2001) Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by β-adrenergic receptor kinase 1. A role in receptor sequestration. J. Biol. Chem., 276, 18953–18959. [DOI] [PubMed] [Google Scholar]
- Nelson G., Chandrashekar,J., Hoon,M.A., Feng,L., Zhao,G., Ryba,N.J. and Zuker,C.S. (2002) An amino-acid taste receptor. Nature, 416, 199–202. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo U.E., Hogue,M., Bhalla,S., Laperriere,A., Morello,J.P. and Bouvier,M. (2002) Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J., 21, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J.A. et al. (1992) Role of βγ subunits of G proteins in targeting the β-adrenergic receptor kinase to membrane-bound receptors. Science, 257, 1264–1267. [DOI] [PubMed] [Google Scholar]
- Pitcher J.A., Freedman,N.J. and Lefkowitz,R.J. (1998a) G protein-coupled receptor kinases. Annu. Rev. Biochem., 67, 653–692. [DOI] [PubMed] [Google Scholar]
- Pitcher J.A. et al. (1998b) The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J. Biol. Chem., 273, 12316–12324. [DOI] [PubMed] [Google Scholar]
- Premont R.T., Inglese,J. and Lefkowitz,R.J. (1995) Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J., 9, 175–182. [DOI] [PubMed] [Google Scholar]
- Premont R.T., Macrae,A.D., Stoffel,R.H., Chung,N., Pitcher,J.A., Ambrose,C., Inglese,J., MacDonald,M.E. and Lefkowitz,R.J. (1996) Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. J. Biol. Chem., 271, 6403–6410. [DOI] [PubMed] [Google Scholar]
- Premont R.T., Claing,A., Vitale,N., Freeman,J.L., Pitcher,J.A., Patton,W.A., Moss,J., Vaughan,M. and Lefkowitz,R.J. (1998) β2-adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc. Natl Acad. Sci. USA, 95, 14082–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallese M., Lombardi,M.S. and De Blasi,A. (1994) Two isoforms of G protein-coupled receptor kinase 4 identified by molecular cloning. Biochem. Biophys. Res. Commun., 199, 848–854. [DOI] [PubMed] [Google Scholar]
- Sallese M., Mariggio,S., Collodel,G., Moretti,E., Piomboni,P., Baccetti,B. and De Blasi,A. (1997) G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. J. Biol. Chem., 272, 10188–10195. [DOI] [PubMed] [Google Scholar]
- Sallese M., Mariggio,S., D’Urbano,E., Iacovelli,L. and De Blasi,A. (2000a) Selective regulation of Gq signaling by G protein-coupled receptor kinase 2: direct interaction of kinase N terminus with activated Gαq. Mol. Pharmacol., 57, 826–831. [PubMed] [Google Scholar]
- Sallese M., Salvatore,L., D’Urbano,E., Sala,G., Storto,M., Launey,T., Nicoletti,F., Knopfel,T. and De Blasi,A. (2000b) The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J., 14, 2569–2580. [DOI] [PubMed] [Google Scholar]
- Shiina T., Arai,K., Tanabe,S., Yoshida,N., Haga,T., Nagao,T. and Kurose,H. (2001) Clathrin box in G protein-coupled receptor kinase 2. J. Biol. Chem., 276, 33019–33026. [DOI] [PubMed] [Google Scholar]
- Usui H. et al. (2000) RGS domain in the amino-terminus of G protein-coupled receptor kinase 2 inhibits Gq-mediated signaling. Int. J. Mol. Med., 5, 335–340. [DOI] [PubMed] [Google Scholar]
- Van Vliet B.J., Sebben,M., Dumuis,A., Gabrion,J., Bockaert,J. and Pin,J.P. (1989) Endogenous amino acid release from cultured cerebellar neuronal cells: effect of tetanus toxin on glutamate release. J. Neurochem., 52, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Virlon B., Firsov,D., Cheval,L., Reiter,E., Troispoux,C., Guillou,F. and Elalouf,J.M. (1998) Rat G protein-coupled receptor kinase GRK4: identification, functional expression and differential tissue distribution of two splice variants. Endocrinology, 139, 2784–2795. [DOI] [PubMed] [Google Scholar]
- Watanabe H., Xu,J., Bengra,C., Jose,P.A. and Felder,R.A. (2002) Desensitization of human renal D1 dopamine receptors by G protein-coupled receptor kinase 4. Kidney Int., 62, 790–798. [DOI] [PubMed] [Google Scholar]
- Weiss E.R., Ducceschi,M.H., Horner,T.J., Li,A., Craft,C.M. and Osawa,S. (2001) Species-specific differences in expression of G-protein-coupled receptor kinase (GRK) 7 and GRK1 in mammalian cone photoreceptor cells: implications for cone cell phototransduction. J. Neurosci., 21, 9175–9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Piston,D.W. and Johnson,C.H. (1999) A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc. Natl Acad. Sci. USA, 96, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]