

Abstract
Although the Cdk inhibitor p21Waf1/Cip1, one of the transcriptional targets of p53, has been implicated in the maintenance of G2 arrest after DNA damage, its function at this stage of the cell cycle is not really understood. Here, we show that the exposure of normal human fibroblasts (NHFs) to genotoxic agents provokes permanent cell cycle exit in G2 phase, whereas mouse embryo fibroblasts and transformed human cells progress through mitosis and arrest in G1 without intervening cytokinesis. p21Waf1/Cip1 exerts a key role in driving this G2 exit both by inhibiting cyclin B1–Cdk1 and cyclin A–Cdk1/2 complexes, which control G2/M progression, and by blocking the phosphorylation of pRb family proteins. NHFs with compromised pRb proteins could still efficiently arrest in G2 but were unable to exit the cell cycle, resulting in cell death. Our experiments show that, when under continuous genotoxic stress, normal cells can reverse their commitment to mitotic progression due to passage through the restriction point and that mechanisms involving p21Waf1/Cip1 and pocket proteins can induce exit in G2 and G1.
Keywords: bleomycin/decatenation/G2 checkpoint/HPV E6 and E7/ICRF-193
Introduction
Cell cycle checkpoints ensure the maintenance of genomic integrity by protecting dividing cells from the potentially fatal consequences of DNA damage. The detection of DNA damage relies on a cascade of enzymes, conveying the signal(s) generated by different genotoxic stresses that blocks key cell cycle transitions until DNA repair has occurred (Abraham, 2001). In the case of irreparable damage, the cells may be forced to withdraw definitively from the cell cycle or die by apoptosis so that cells do not replicate or segregate chromosomes bearing unrepaired lesions. Defects in the DNA damage checkpoint and/or the related cell cycle regulation network could contribute to the development of diverse types of mutations or chromosome rearrangements and promote tumorigenesis.
After DNA damage, normal cells arrest at the G1/S or G2/M transitions of the cell cycle. Cells lacking p53, the most commonly mutated tumour suppressor gene, are defective for the G1 checkpoint but retain the ability to arrest (at least temporarily) in G2 (Lowe et al., 1993; Bunz et al., 1998). This arrest is thought to occur by mechanisms that maintain inhibitory phosphorylation on Thr14 and Tyr15 of Cdk1 by preventing Cdc25C activation (Peng et al., 1997) and/or blocking nuclear accumulation of cyclin B1–Cdk1 complexes (Yang et al., 1998).
p53 is stabilized in response to genotoxic stress and controls the upregulation of several genes implicated in both the G1/S and G2/M transitions (Taylor and Stark, 2001). Among them is the Cdk inhibitor p21Waf1/Cip1 (hereafter referred to as p21), a well-established negative regulator of the G1/S transition (Sherr and Roberts, 1999) whose role in G2 arrest has been documented by several studies (Bunz et al., 1998; Taylor and Stark, 2001). The prevailing view is that p21 is not essential for initiating G2 arrest but rather contributes to its maintenance. Although widely used, the exact meaning of this concept has remained unclear.
The precise role and critical G2 targets of p21 in response to DNA damage have not yet been defined, partly due to the use of human tumour-derived cell lines, whose checkpoint pathways are not always intact. Because of its low affinity towards Cdk1, it is thought that p21 plays a rather marginal role in its inactivation. Even though it has been reported that ectopic induction of p21 inhibits cyclin B1-associated kinase activity by blocking Thr161 phosphorylation of Cdk1, in general, only a minor fraction of cyclin B1–Cdk1 complexes is found to be associated with p21 (Winters et al., 1998; Smits et al., 2000). Other likely targets of p21 in G2 are cyclin A–Cdk1/2 complexes (Dulić et al., 1998; Smits et al., 2000), which also control mitotic entry (Pagano et al., 1992; Furuno et al., 1999; Hu et al., 2001). Nevertheless, the importance of their inactivation for G2 arrest has not been thoroughly addressed.
We investigated the role of p21 in the regulation of G2 arrest and its maintenance in response to DNA damage. To ensure maximal integrity of checkpoints, we chose as a model normal human fibroblasts (NHFs) and mouse embryo fibroblasts (MEFs) that were exposed to two genotoxic agents exhibiting different properties of action, ICRF-193 and bleomycin. ICRF-193 is a topoisomerase II inhibitor, which provokes G2 arrest or delay (Ishida et al., 1994) without causing detectable DNA double-strand breaks (DSBs) (Downes et al., 1994; Kaufmann and Kies, 1998; Deming et al., 2001). Bleomycin is a radiomimetic drug causing DSBs that, when added to cycling cells, blocks both the G1/S and G2/M transitions (Kaufmann and Kies, 1998).
We show that, in response to a chronic genotoxic stress leading to G2 arrest, NHFs, but not MEFs nor transformed cell lines, are able to exit from the cell cycle into a quiescence-like state, thus preventing entry into mitosis. Furthermore, from experiments using NHFs whose p53 or pocket proteins are inactivated by viral oncogenes, we found that p21 probably has a double role in this process. First, it contributes to G2 arrest by the inactivation of cyclin A–Cdk1/2 and a subset of cyclin B1–Cdk1 complexes. Secondly, as described for the G1 arrest following DNA damage, p21 in G2 inhibits Cdks involved in the hyperphosphorylation and inactivation of pocket proteins of the retinoblastoma family. We propose that the resulting accumulation of active pocket proteins is absolutely required to drive the cell cycle exit.
Results
MEFs are deficient in blocking mitosis in response to bleomycin and ICRF-193
To investigate the mechanisms of G2 arrest in response to ICRF-193 and bleomycin, we initially chose NHFs and MEFs as models. We purposely did not use experimental systems based on tumour-derived cells lines, as many of them have acquired uncharacterized genetic abnormalities, which can confound the interpretation of the data obtained. Moreover, the cell lines we tested exhibited aberrant post-mitotic figures suggesting that their G2/M checkpoint is not entirely functional (data not shown). The ability of asynchronously growing NHFs and MEFs to progress into mitosis in the continuous presence of each drug was monitored for 24 or 48 h by video-microscopy (data now shown). In addition, we carried out microscopic examination of Hoechst-stained fixed nuclei.
After the addition of drugs, very few mitotic cells were observed in NHF cultures, but, to our surprise, we found that MEFs behaved as if their G2/M checkpoint was non-functional, since in the presence of either drug they readily entered mitosis (Figure 1A): both mitotic cells and aberrant post-mitotic nuclei (Figure 1) were evident. Moreover, the drug-treated MEF cultures contained significant numbers of dying cells (data not shown). In contrast, only a small fraction of abnormal bi-nucleated cells was observed after ICRF-193 treatment of NHFs. These may arise as a consequence of cell leakage through the decatenation checkpoint or more likely represent cells trapped by ICRF-193 in mitosis (Ishida et al., 1994). In synchronized NHFs treated with ICRF-193, mitoses and aberrant post-mitotic figures were extremely rare (see Supplementary figure 1 available at The EMBO Journal Online). G2 arrest induced by bleomycin was even more efficient, as judged by the absence of mitotic cells and by the presence of few abnormal post-mitotic nuclei (Figure 1A). We conclude that MEFs are not the best model for studying the G2/M transition after DNA damage, as they failed to block mitotic entry when treated with agents known to activate a checkpoint-dependent G2 arrest.

Fig. 1. Mouse embryo fibroblasts (MEFs) have a non-functional G2/M checkpoint in comparison with normal human fibroblasts (NHFs). (A) Percentage of cells in mitosis or with aberrant post-mitotic nuclei (PMN) in untreated (Ct) and 24 h ICRF-193-treated (Ic) and bleomycin-treated (Bl) NHF and MEF cultures. For each situation, at least 500 cells were scored. (B) Micrographs (Hoechst) showing untreated MEFs (Cont) and some characteristic bleomycin- and ICRF-193-induced mitotic and aberrant PMN (arrowheads) after 24 h exposure. Arrows point to normal mitoses in untreated cells.
Bleomycin and ICRF-193 induce rapid association of p21 with Cdks controlling the G2/M transition
To assess the effect of bleomycin and ICRF-193 on cell cycle progression, we initially studied asynchronously growing NHFs exposed to these drugs for different times. As shown by flow cytometric analysis, NHFs exposed to ICRF-193 specifically accumulated in G2, whereas, as expected, bleomycin induced both G1 and G2 arrest (Figure 2A). Both genotoxic agents induced a rapid accumulation of p53 and p21, which were readily detected after 3 h of treatment (Figure 2B). Western blot analysis of p21 immunoprecipitates showed that, in response to both drugs, p21 increasingly associates with cyclin A and cyclin B1 and with cognate kinases Cdk2 and Cdk1 (Figure 2C). Note that p21 is equally bound to hypophosphorylated and hyperphosphorylated Cdk1 and Cdk2 isoforms, suggesting that its presence interferes with both Cdk phosphorylation and dephosphorylation.

Fig. 2. In response to DNA damage, p21 targets Cdks regulating the G2/M transition. (A) Cell cycle profiles of exponentially growing cells exposed to ICRF-193 and bleomycin for the indicated times. Percentage of cells containing 4N DNA content is shown on the right of each FACS profile. (B) p53 and p21 protein levels in untreated cells (Ct) or cells exposed to ICRF-193 (Ic) or bleomycin (Bl) for the indicated times. (C) Western blot analysis of p21 immunoprecipitates (IPs) isolated from control cells and those treated for 12 h with ICRF-193 or bleomycin. Cdk1 migrates as three bands (1–3), whereas Cdk2 migrates as two bands (1 and 2). Number 1 points to CAK-phosphorylated/Cdc25-dephosphorylated Cdk isoforms (potentially active). Note that antibody directed against phospho-Thr161 (P-T161) recognizes both Cdk1 and Cdk2 (asterisk). (D) Immunoblot analysis of cyclin complexes isolated before and after p21 immunodepletion. Cell lysates prepared from untreated cells and those treated for 12 h (as above) were immunodepleted for p21-associated complexes by successive incubations with an anti-p21 antibody (+). Mock samples of the same extracts were incubated with protein A beads alone (-). Cyclin IPs were examined for cyclin, Cdk and p21 content. Numbers 1–3 point to the different Cdk1 and Cdk2 isoforms. In cyclin B1 IP, the arrow points to the hypophosphorylated Cdk1 (1) isoform. Note that antibody directed against P-T161 recognizes both Cdk1 and Cdk2 (asterisk).
To estimate which subpopulation of cyclin–Cdk complexes controlling G2/M progression is being targeted by p21, and to what extent, we analysed cyclin B1 and cyclin A immunoprecipitates isolated before (–) and after (+) the removal of p21-bound complexes by immunodepletion. As shown in Figure 2D, the vast majority of cyclin A–Cdk1/2 complexes, accumulated in the presence of both drugs (12 h), was associated with p21. In the case of cyclin B1–Cdk1 complexes, drug-induced association with p21 was significant but less quantitative, and p21 appears to bind to both hyperphosphorylated (isoform 3) and hypophosphorylated (isoform 1) Cdk1 (Figure 2D). However, cyclin B1-associated Cdk1 isoform 1 removed by p21 was not recognized by the antibody directed against phospho-Thr161 (P-T161), suggesting that p21 inhibits CAK-mediated phosphorylation of this residue, as proposed earlier by Smits et al. (2000).
Next, we wished to determine whether the association of mitotic cyclin–Cdk complexes with p21 might account for their inactivation leading to G2 arrest. To this end, we studied NHFs synchronized at the G1/S boundary, followed by re-entry into the cell cycle in the presence, or not, of ICRF-193 and bleomycin. The progression into the cell cycle was monitored by flow cytometry in order to determine DNA content and entry into mitosis through the detection of cells labelled with antibodies directed against phospho-histone H3 (Figure 3A). The untreated cells were harvested at different times corresponding to mid-S phase (3 h), G2 phase (7 h) and the G2/M transition (9.5 h). The drug-treated cells were harvested at the time when the majority of control cells entered mitosis. Indeed, mitosis-associated increase of cyclin A- and B1-associated histone H1 kinase activity and cyclin autophosphorylation were efficiently blocked by both drugs, even in the presence of nocodazole, which was added to ‘trap’ the cells in mitosis (Figure 3B).

Fig. 3. Bleomycin and ICRF-193 block mitosis by preventing activation of mitotic cyclin–Cdk complexes. (A) Flow cytometry analysis of normal human fibroblasts synchronized at the G1/S transition and released into the cell cycle in the absence or presence of bleomycin and ICRF-193. Mitotic cells were labelled by using the antibody directed against phospho-histone H3 (black-shaded profile). Untreated cells were harvested and analysed at 3 h (S phase), 7 h (G2 phase) and 9.5 h (G2/M transition) after release from the G1/S block. ICRF-193- and bleomycin-treated samples were collected at the time when most control cells have entered mitosis (G2/M), 2 h later in the presence of nocodazole (Noco), and after 24 h. (B) Histone H1 kinase (H1) activity and cyclin autophosphorylation of cyclin B1 (cB1) and cyclin A (cA) immunoprecipitates (IPs) isolated from corresponding cell extracts. Noco denotes the cultures to which nocodazole was added (2 h) to ‘trap’ the cells in metaphase. (C) Western blot analysis of cyclin B1 and cyclin A IPs isolated from synchronized untreated and drug-treated cells showing cyclin, Cdk and p21 content. In cyclin B1 immunoblots, arrows point to the hypophosphorylated cyclin B1-associated Cdk1 band that specifically accumulates in mitotis. In drug-treated cells, the Cdk1 band migrating at the same position, but not recognized by an antibody directed against phospho-Thr161 (P-T161), is removed by p21 immunodepletion (cf. Figures 2D and 6C). Note that the lowest band (asterisk) in cyclin A IP is composed of hyperphosphorylated Cdk2 and hypophosphorylated Cdk1 isoforms, as anti-P-T161 antibody recognizes both Cdks. Numbered arrows point to the different Cdk isoforms.
Molecular mechanisms underlying Cdk inactivation were assessed by immunoblot analysis of corresponding cyclin immunoprecipitates (Figure 3C). In the case of cyclin B1–Cdk1 complexes, inactivation appears to be accomplished predominantly, but not exclusively, by the inhibition of Cdc25 phosphatase. This was documented by the near-absence of hypophosphorylated (P-T161 positive) Cdk1 in cyclin B1 complexes (isoforms 1 and 2) in G2-arrested cells. Nevertheless, and in agreement with the results obtained in asynchronous cells (Figure 2C and D), the drug treatment also induced an accumulation of p21–cyclin B1 complexes. The significance of p21 in inactivating cyclin B1–Cdk1 complexes is currently under investigation. Western blot analysis of cyclin A–Cdk1/2 complexes confirmed that their inactivation is mostly due to association with p21, as Cdk phosphorylation status (detected by anti-P-T161 antibody) is only slightly affected in the presence of either drug. Note the apparent absence of Cdk1 isoform 2 (the intermediate phosphorylated state).
The above results strongly suggest that, in NHFs, p21 regulates the activity of Cdks involved in G2/M progression in response to genotoxic stress. Although cyclin B1–Cdk1 complexes also contain p21, the major targets of this inhibitor appear to be cyclin A–Cdk1/2 complexes. Thus, these results are consistent with two potential scenarios in which p21 could have a role either in G2 arrest or in its maintenance (Bunz et al., 1998).
Role of p53/p21 pathway in DNA damage-induced G2 arrest
To distinguish between these two possible roles of p21, we studied NHFs expressing the HPV-16 E6 oncogene (hereafter referred to as E6 cells) that, owing to the ability of E6 oncoprotein to target p53 for degradation (Scheffner et al., 1993), contain very low amounts of p21 (Figure 4A). Although E6 oncoprotein has targets other than p53 (Mantovani and Banks, 2001), E6 cells are commonly used as a model of p53-deficient cells. p21–/– MEFs were not used for these studies, since some G2/M checkpoints seem to be absent even in wild-type MEFs (Figure 1; Borel et al., 2002).

Fig. 4. Impaired but differential response to ICRF-193 and bleomycin in normal human fibroblasts (NHFs) deficient for p53/p21 pathway. (A) p53 and p21 proteins levels in untreated (Ct), ICRF-193-treated (Ic) and bleomycin-treated (Bl) wild-type (WT) and E6 NHFs. Note that the low level of p53 detected in E6 cells did not increase after drug treatment. (B) Cell cycle profiles of WT and E6 NHFs exposed to ICRF-193 or bleomycin for 24 h. (C) Histone H1 kinase activity and western blot (WB) analysis of cyclin B1 and cyclin A immunoprecipitates (IPs) isolated from untreated (Ct), ICRF-193- and bleomycin-treated WT and E6 cells. Numbers 1–3 point to the different Cdk isoforms. (D) Flow cytometry analysis (FACS) of E6 cells synchronized at G1/S boundary and released into the cell cycle in the absence (Cont) or presence of the indicated drug. Mitotic cells were labelled by using the antibody directed against phospho-histone H3 (black-shaded profile). Untreated cells were harvested at 6 h (G2 phase) and 8 h (G2/M transition) and in the presence of nocodazole (8 + 2 h). Drug-treated cells were harvested 10 and 24 h after release from the block. (E) Histone H1 kinase activity and WB analysis of cyclin B1 and cyclin A IPs isolated from synchronized untreated (Ct) and ICRF-193- and bleomycin-treated E6 cells. Extracts were analysed from the cells harvested at the indicated times after release from the G1/S block (cf. the FACS analysis above). Arrows point to the hypophosphorylated cyclin B1-associated Cdk1 (isoform 1), which is absent in S phase and enriched in mitotic cells. In the cyclin A IPs, the lowest PSTAIRE-recognized band (1) consists of both hypophosphorylated Cdk1 and CAK-phosphorylated Cdk2 isoforms. Numbers 1–3 point to the different Cdk isoforms. (F) Micrographs taken from a video-microscopy sequence showing E6 cells in the absence (Ct) of the drugs and those exposed for 72 h to bleomycin or ICRF-193. Untreated cells were shown at the beginning of the experiment (0 h; at the time when the drugs were added) and after 72 h. Note the dying cells in drug-treated cultures.
Initially, we examined the effect of bleomycin and ICRF-193 on asynchronously growing E6 cells. In contrast to wild-type cells, the presence of these drugs did not induce p53 stabilization and accumulation of p21 (Figure 4A). Interestingly, FACS analysis of DNA content and histone H3 phosphorylation revealed that, whereas ICRF-193-treated E6 cells accumulated with 4N DNA content including a large number of mitotic cells, bleomycin-treated cells arrested mainly in S and G2 phases (Figure 4B and D). This differential response to two drugs was further confirmed by microscopic observation of cell cultures and biochemical analysis of cyclin immunoprecipitates isolated from corresponding cell extracts. Cyclin B1 immunoprecipitates from ICRF-193-treated cells exhibited extremely high kinase activity when compared with control and bleomycin-treated cells (Figure 4C). Note that, in the latter, neither cyclin A- nor cyclin B1-associated kinases were efficiently inactivated.
Western blot analysis of cyclin immunoprecipitates indicated that bleomycin blocked dephosphorylation/ activation of Cdk1, as no hypophosphorylated Cdk1 (isoform 1) could be detected with cyclin B1. In contrast, in ICRF-193-treated cells, cyclin B1 complexes contained a substantial fraction of hypophosphorylated and active Cdk1 (isoform 1 in Figure 4C). These results implied that, in E6 cells, the decatenation checkpoint is either absent or that maintenance of G2 arrest is inefficient.
To distinguish between these two possibilities, we monitored synchronized E6 cells exposed to ICRF-193 or bleomycin by video-microscopy. At the time when untreated cells entered mitosis, mitotic cells were absent in the presence of either drug, indicating that the p53/p21 pathway is not required for initiating G2 arrest. However, after a brief delay (3–4 h), increasing numbers of mitotic cells were observed in the presence of ICRF-193, whereas very few could be seen in bleomycin-treated cultures (see Supplementary figure 4). The existing difference in response to these drugs was further strengthened by flow cytometry analysis, which showed differential cell cycle profiles as well as the absence of phospho-histone H3 in drug-arrested cultures (Figure 4D). Moreover, in agreement with video-microscopy results, ICRF-193-treated cultures contained almost as many mitotic cells after 24 h as those exposed to nocodazole.
These observations were further corroborated by the results obtained after examination of cyclin B1 and cyclin A immunoprecipitates isolated from synchronized cells. At the time when untreated cells entered mitosis, exhibiting increased cyclin B1-associated kinase activity, in drug-treated cells this kinase was inactive and cyclin B1 complexes lacked hypophosphorylated Cdk1 (Figure 4E). In contrast, 24 h after the release from G1/S block, cyclin B1-associated kinase activity was dramatically elevated in the presence of ICRF-193 and was comparable with nocodazole-treated control cells. This correlated with an accumulation of hypophosphorylated Cdk1 (Figure 4E), indicating that Cdc25 phosphatase became active. As expected from video-microscopy data and the flow cytometry profiles, cyclin B1-associated kinase activity was much weaker in bleomycin-treated E6 cells due to the presence of mostly hyperphosphorylated Cdk1 (isoform 3 in Figure 4E). In comparison with cyclin B1–Cdk1 complexes, cyclin A-associated kinase was less affected by these drugs (Figure 4E) and its activity remained elevated.
These results implied that the p53/p21 pathway is not essential for G2 arrest in response to bleomycin but that it is required for full implementation (i.e. arrest and maintenance) of the decatenation checkpoint.
Continuous genotoxic stress in G2 leads to an irreversible cell cycle exit
The above results could not entirely explain the functional significance of the massive association of p21 with cyclin A–Cdk1/2 complexes observed in NHFs in response to DNA damage. We postulated that p21-mediated Cdk inactivation might be required not only to block mitotic events but also to maintain the G2 arrest and drive cell cycle exit, for the following reasons. First, cyclin A-associated kinase remained active in E6 cells exposed to both drugs even in the absence of mitotic events (Figures 4E). Secondly, after prolonged exposure (72 h) to either drug, E6 cells eventually started to die (Figure 4F), while NHFs remained in a viable arrest (see below). Whereas, in ICRF-193-treated E6 cultures, cell death was presumably provoked by the lethal consequences of aberrant mitoses, this explanation is probably not valid in the case of cells treated with bleomycin, because most of them fail to undergo mitosis (Figure 4D).
In G1, cell cycle exit is mediated by the inactivation of Cdks that control the initiation of S phase and are involved in the phosphorylation of the pocket proteins of the pRb family (pRb, p130 and p107). In their active hypophosphorylated state, pocket proteins prevent G1/S cell cycle progression by sequestering transcription factors of the E2F family and by blocking directly the expression of genes required for the initiation of S phase (Grana et al., 1998). We speculated that continuous genotoxic stress in G2 could, via p21-dependent inactivation of cyclin–Cdk complexes, block the phosphorylation of pocket proteins, thereby initiating cell cycle exit before mitosis. This model implies that phosphorylation of pocket proteins is required for G2/M progression.
To explore this possibility, we examined whether the treatment of asynchronously growing NHFs with ICRF-193, which leads to G2 arrest, affects the phosphorylation of pocket proteins. Hyperphosphorylation of pRb, p107 and p130 was blocked very early (6 h) after exposure to ICRF-193 or bleomycin (Figure 5A), concomitant with an increase of p21 and the appearance of cells with 4N DNA content (Figure 2A). Moreover, 24 h after the addition of the drugs, arrested cells behaved in many aspects as quiescent (G0) cells, i.e. they accumulated hypophosphorylated (active) forms of p130 and pRb, whereas p107 levels strongly diminished, lacked both S phase and mitotic cyclin–Cdk complexes and, in addition to p21, contained elevated amounts of p27Kip1 (Figure 5A).

Fig. 5. DNA damage leads to irreversible cell cycle exit in G2. (A) Western blot analysis of protein lysates prepared from exponentially growing normal human fibroblasts (NHFs) untreated (Ct) or exposed at various times to ICRF-193 (Ic) and bleomycin (Bl). G0 denotes contact inhibited NHFs. To assess different aspects of regulation contributing to cell cycle arrest and exit, levels of pocket proteins (pRb, p130 and p107), Cdk inhibitors p21 and p27, and cyclin A were monitored. Note the rapid accumulation of hypophosphorylated forms of pocket proteins (arrows) while cells still contained high levels of cyclin A. (B) Western blot analysis showing phosphorylation status of pocket proteins in control and drug-treated synchronized NHFs. FACS profile of cells exposed to ICRF-193 and bleomycin collected 24 h after the G1/S block is shown in Figure 3A. Biochemical analysis of corresponding cyclin immunoprecipitates is shown in Figure 3A and B. (C) Western blot analysis showing pRb and mitotic cyclin levels in control and drug-treated synchronized E6 cells. The same samples are shown in Figure 4. Chk2 status was monitored to assess efficient signalling in response to DNA damage in E6 cells. (D) Cell size analysis (flow cytometry) of control G2 cells versus G2 cells that were exposed to ICRF-193 or bleomycin for 7 and 24 h. (E) Micrographs showing cell morphology and β-gal staining of untreated NHFs (a) and cultures exposed to ICRF-193 for 24 h (b) and 72 h (c) after treatment. As positive controls, NHFs expressing oncogenic ras (d) and senescent culture (e) are shown.
To demonstrate that the hypophosphorylation of pocket proteins occurred specifically in G2, their status was examined in the synchronized cells to which the drugs were added after a release from the G1/S boundary (Figure 3). As shown in Figure 5B, this was the case. Moreover, 24 h after the addition of drugs, pocket proteins became completely hypophosphorylated, whereas amounts of mitotic cyclins severely diminished even though virtually all cells exhibited a 4N DNA content (cf. Figure 3A). To ascertain that these cells did not undergo mitosis without cytokinesis and instead arrested in a 4N tetraploid state, like MEFs (cf. Figure 1), the cultures were examined by microscopy and video-microscopy. No such events were observed (data not shown).
A corollary of these results was that p21 inactivates Cdk implicated in the phosphorylation of pocket proteins also in S and G2 phases. This idea was further supported by our results showing that, in synchronized E6 cells, pRb phosphorylation was not inhibited even under prolonged exposure to either drug (Figure 5C). In addition, our finding that both bleomycin and ICRF-193 induced Chk2 phosphorylation showed that the DNA damage signalling pathway in E6 cells is intact (Figure 5C). Interestingly, this phosphorylation is only partial in response to ICRF-193, which may explain the inefficient maintenance of G2 arrest observed in the cells exposed to this drug.
The absence of cyclin A and cyclin B, the accumulation of hypophosphorylated pocket proteins and the steady increase of p27 suggested that this G2 exit from the cell cycle was a point without return. We confirmed this notion by showing that, after drug removal, the cells were unable to re-initiate the cell cycle (measured by BrdU incorporation) even after a recovery period of 6 days (see Supplementary figure 5). Moreover, in continuous presence of both drugs (or even when the drugs were removed and the cells were left for several days), the cells became large, resembling senescent cells (Figure 5D). However, unlike NHFs whose proliferation was blocked due to replicative senescence or by overexpression of oncogenic ras, the cells that exited the cell cycle in G2 (ICRF-193) did not exhibit significant β-gal staining (Figure 5E).
Disruption of pocket protein function abrogates cell cycle exit in G2
If active pocket proteins were required for the cell cycle exit in G2, the cells lacking active pocket proteins but containing active p53 should efficiently block entry into mitosis, but the G2 arrest alone would not be sufficient to drive cell cycle exit. To test this hypothesis, we studied isogenic NHFs expressing the HPV-16 E7 oncogene (hereafter referred to as E7 cells), which inactivates pocket proteins (Munger et al., 2001). Although E7 cells are deficient in radiation-induced G1 arrest in spite of the induction of p53 and p21, they largely retain functional mitotic checkpoints (Munger et al., 2001; and references therein).
Figure 6A shows a comparison of the status of pocket proteins (pRB, p130 and p107) in asynchronous wild-type, E6 and E7 cells exposed to ICRF-193 and bleomycin. As expected, and unlike wild-type cells, E6 cells failed to block the hyperphosphorylation of pRb and p107 and were deficient in the accumulation of hypophosphorylated p130. It should be noted that, in the absence of growth factors or when confluent, E6 cells were perfectly capable of arresting in G0 with hypophosphorylated pocket proteins (Dulić et al., 2000; data not shown). In contrast to wild-type and E6 cells, E7 cells contained very low amounts of pocket proteins, but their DNA damage checkpoint appeared to be intact: both drugs activated p53, resulting in significant induction of p21 levels (Figure 6A), and efficiently blocked G2/M progression, as revealed by video-microscopy and immunofluorescence microscopy (data not shown). This notion was further supported by biochemical analysis of cyclin immunoprecipitates in synchronized cells, which showed that mitosis-specific activation of both cyclin A and cyclin B1 complexes was strongly inhibited in the presence of either drug (Figure 6B).

Fig. 6. Active pocket proteins are required for cell cycle exit in G2 after genotoxic stress. (A) Western blot analysis of protein extracts prepared from exponentially growing wild-type (WT) and E6- and E7-expressing normal human fibroblasts (NHFs) that were untreated (Ct) or exposed to ICRF-193 (Ic) and bleomycin (Bl) for 24 h. Note the persistent high levels of cyclin A in drug-treated E6 and E7 cells. (B) Histone H1 kinase activity and western blot (WB) analysis of cyclin B1 and cyclin A immunoprecipitates (IPs) isolated from synchronized untreated, ICRF-193- and bleomycin-treated E7 fibroblasts. Untreated cells were harvested in S, G2 and M phases. Nocodazole was added to both control and treated cells to ‘trap’ mitotic cells. Autoradiographs show both histone H1 and autophosphorylated cyclins. To accentuate the increase cyclin A-associated kinase activity in mitosis, both short and long (*H1) autoradiograph exposure are shown. The same immunoblots were analysed for the presence of cyclins (A and B1) and Cdk. Numbered arrows point to the different Cdk1 phospho- isoforms. (C) Kinase activity and WB analysis of cyclin B1 IPs isolated from untreated (Ct), ICRF-193- and bleomycin-treated (6 h) asynchronously growing WT and E7 cells. Numbered arrows point to the hyperphosphorylated (3) and hypophosphorylated (1) cyclin B1-associated Cdk1 isoforms. Note that the latter isoform does not appear to be phosphorylated at T161 in G2-arrested cells. (D) Prolonged exposure to ICRF-193 and bleomycin provokes cell death in spite of efficient G2 arrest. The micrographs (extracted from a video-microscopy sequence) show control and ICRF-193- and bleomycin-treated cultures after 48 h.
Surprisingly, in spite of their inactive state, in arrested cells cyclin B1 complexes contained high amounts of hypophosphorylated Cdk1 (isoform 1) identified as an active form. This Cdk isoform, albeit much less abundant, was also observed in inactive cyclin B complexes of G2-arrested wild-type NHFs, but, in contrast to active Cdk1, it was not phosphorylated on T161 (Figure 2D). Therefore, we assumed that inactivity of these complexes is partly due to association with p21. Western blot analysis of cyclin B1 immunoprecipitates from drug-treated E7 cells showed a dramatic accumulation of this isoform concomitant with increasing association with p21 (Figure 6C). As in wild-type cells, this form is not phosphorylated on T161 (in contrast to isoform 3; Figure 6C) and is removed by p21 immunodepletion (F.Baus, V.Gire, J.Piette and V.Dulić, in preparation).
However, in spite of their efficient G2 arrest in response to both ICRF-193 and bleomycin, prolonged exposure to either drug resulted in a massive cell death of E7 cells. This effect was even more accentuated in the case of bleomycin (Figure 6D). We conclude that cell death was a consequence of the absence of active pocket proteins, which appear to be required for the initiation of necessary events leading to G2 cell cycle exit.
Discussion
We describe a mechanism by which, in response to chronic exposure to genotoxic agents, NHFs permanently exit from the cell cycle in G2, thus efficiently blocking entry into mitosis and avoiding genetic instability. Our experiments argue strongly that this G2 exit, in many aspects resembling a quiescent state, is initiated a few hours after exposure to genotoxic drugs and that its execution depends on the integrity of both p53 and pocket proteins of the Rb family. A similar biological situation, i.e. p53-dependent cell cycle exit with 4N DNA content after DNA damage, has recently been reported by others (Stewart et al., 1999; Andreassen et al., 2001a), although the biochemical mechanism was not described. However, an important difference lies in the fact that the immortal cell lines (such as HCT116) used in these and other studies, after an initial G2 delay, eventually progressed into mitosis and, in the absence of cytokinesis, arrested as tetraploid cells containing two G1 nuclei (Andreassen et al., 2001b). Unexpectedly, we found that, in the presence of the radiomimetic drug bleomycin or topoisomerase II inhibitor ICRF-193, MEFs were also able to undergo mitosis, giving rise to numerous aberrant two-nuclei figures. This was in striking contrast to NHFs, where such situations are rarely observed (Kaufmann and Kies, 1998; Deming et al., 2001). These and other observations (Borel et al., 2002) suggested that, unlike in NHFs, some G2 checkpoints are non-functional in MEFs and in tumour-derived cell lines [e.g. RKO (Flatt et al., 2000; Gottifredi et al., 2001)], therefore emphasizing the importance of studying genotoxic stress responses in normal human cells.
The original aim of the study was to understand the role of p21 in the G2 arrest in response to DNA damage. Although the role of p21 in sustaining G2 arrest had been proposed earlier (Agarwal et al., 1995; Bunz et al., 1998; Ohi and Gould, 1999), the mechanisms whereby this process is accomplished were not addressed. Several other papers have described experiments addressing these questions (see below), but the interpretation of the results may be hampered by the fact that these studies were mostly carried out in immortalized human cell lines.
In the context of a permanent G2 exit, we show that, in NHFs, p21 plays a key role by interfering with at least two apparently separate processes (Figure 7). The first is the regulation of the G2/M transition, in which p21 may participate directly by inhibiting mitotic cyclin–Cdk complexes, and the second is the phosphorylation of pocket proteins of the pRb family.
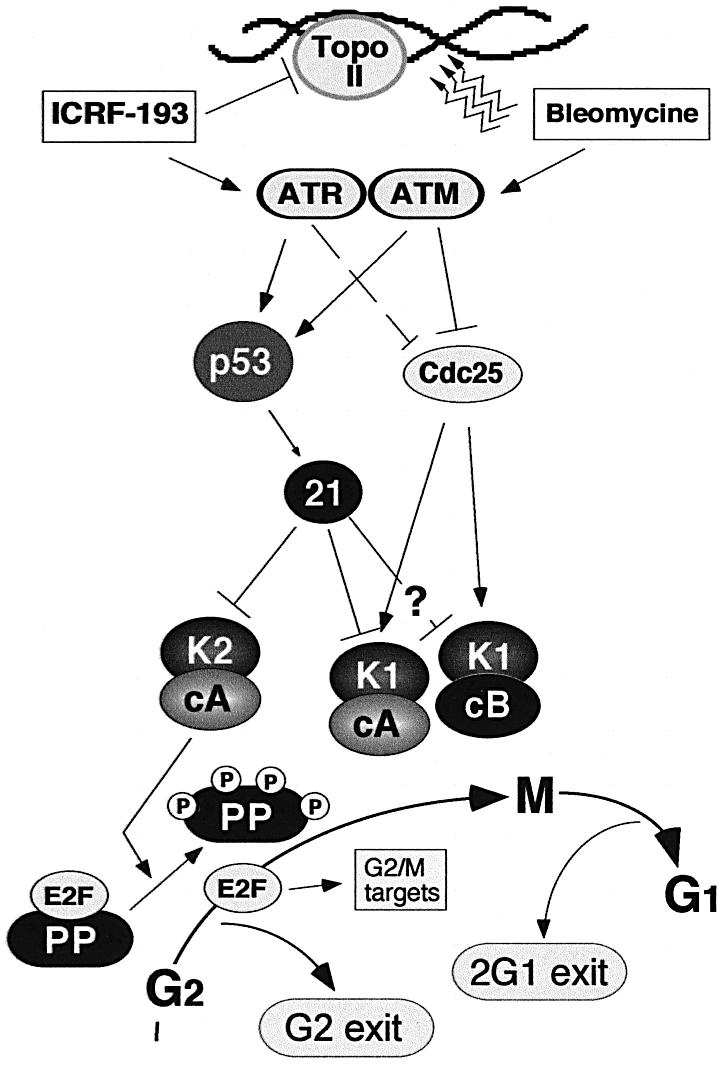
Fig. 7. Role of p21 in directing cell cycle exit after DNA damage in G2. In response to DNA damage (bleomycin) and the inhibition of topoisomerase II (ICRF-193), ATM and ATR kinases, respectively, are activated (Deming et al., 2001). In E6 cells, the inactivation of Cdc25 via the ATR pathway by ICRF-193 is less efficient. Cell cycle arrest is accomplished by a network of enzymes that block activation of the mitotic kinases cyclin A–Cdk1 and cyclin B1–Cdk1. The question mark refers to the fact that the role of p21 in inactivating cyclin B1–Cdk1 complexes is less well established. In addition to blocking activation of mitotic kinases, p21 also inactivates Cdks involved in the hyperphosphorylation of pocket proteins (PP) of the retinoblastoma family (pRb, p107 and p130) leading to the accumulation of hypophosphorylated (active) pocket proteins. The resulting sequestration of transcription factors of the E2F family would block transcription of a number of genes involved in G2/M progression (Taylor et al., 2001; Ren et al., 2002), thereby driving cell cycle exit in G2. In mouse fibroblasts and tumour-derived cell lines, the exit is accomplished only after mitosis but before cytokinesis, giving rise to tetraploid cells (2G1 exit; cf. Borel et al., 2002). The model also shows that, in the case of ICRF-193-induced G2 arrest, the inactivation of Cdc25 pathways is less efficient than that provoked by double-strand DNA breaks (bleomycin).
This first role is supported by our finding that virtually all cyclin A–Cdk1/2 complexes were associated with p21 after exposure to all genotoxic drugs tested. Since, in addition to cyclin B1–Cdk1, both cyclin A–Cdk2 (Hu et al., 2001) and cyclin A–Cdk1 (Furuno et al., 1999) complexes have been shown to play an important role in G2/M progression, their inactivation by p21 may therefore influence mitotic entry directly. However, our data showing that p21 binds only to a relatively minor subpopulation of cyclin B1–Cdk1 complexes, which is in agreement with other reports (Flatt et al., 2000; Smits et al., 2000; Deming et al., 2001), imply that it is the Cdc25 pathway that plays a major role in controlling their activity. Indeed, in E6 cells, which contain low p21 levels, both drugs prevented timely activation of cyclin B1–Cdk1 complexes and blocked entry into mitosis. However, whereas bleomycin-mediated G2 arrest appears to be sustained (Passalaris et al., 1999; Flatt et al., 2000), ICRF-193-treated cells eventually accumulated in mitosis and often gave rise to aberrant post-mitotic figures. Apparent differences in cellular responses to decatenation and DSB checkpoints (Deming et al., 2001), which are also reflected in different cell cycle profiles, may originate in unequal degrees of Chk2 activation. Nevertheless, taking into account the significant amount of p21-bound cyclin B1–Cdk1 complexes in drug-treated E7 cells, it is possible that, in response to DNA damage, p21-dependent inactivation of cyclin–Cdk1 complexes may function in synergy with the Cdc25 pathway. We are currently studying the role of p21 in the inactivation of cyclin B1–Cdk1 complexes.
A second, and hitherto undescribed, role of p21 in DNA damage-induced G2 exit is to block the hyperphosphorylation of pocket proteins (pRb, p107 and p130) occurring in post-G1 phases of the cell cycle via the inactivation of cyclin A–Cdk (and perhaps other Cdk) complexes. The resulting accumulation of active pocket proteins would, in turn, drive cell cycle exit in G2 by sequestering the members of the E2F transcription factor family, which control expression of a number of genes involved in G2/M progression (cf. Taylor et al., 2001; Ren et al., 2002). An untimely activation of these genes would lead to cell death, possibly due to contradictory growth signals. Several lines of evidence, described below, support this hypothesis.
In NHFs whose p53/p21 pathway was compromised by expression of the HPV-16 E6 oncogene, hyperphosphorylation of pocket proteins did not diminish in the presence of either bleomycin or ICRF-193, and cyclin A-associated kinase was active throughout treatment. These results were consistent with the role of p21 in the inactivation of cyclin A–Cdk complexes involved in the phosphorylation of pocket proteins (Figure 7). Although both drugs could, at least temporarily, block G2/M progression, the cells are unable to exit the cell cycle and either progress toward mitosis and eventually die due to a mitotic catastrophy (ICRF-193) or start dying without entering mitosis.
As opposed to E6 cells, NHFs whose pocket proteins were inactivated by the expression of the HPV-16 E7 oncogene, but contain an intact p53/p21 pathway, were able to efficiently inactivate both cyclin A–Cdk and cyclin B1–Cdk complexes and readily blocked mitosis in response to either drug. Moreover, we show that these complexes are inactivated in E7 cells, at least in part, by association with p21, thus arguing against a prevailing view that the E7 oncogene, by binding to p21, abrogates its inhibitory function (Munger et al., 2001). Nevertheless, G2 arrest by itself was insufficient to confer cell cycle exit, and cell death ensued after prolonged exposure to the drugs. Moreover, these observations may account for important differences between our results and those reported by others in which the cells with inactive pocket proteins underwent re-replication in response to DNA damage (Flatt et al., 2000). The fact that cyclin–Cdk complexes were not inactivated in RKO-E7 cells in response to DNA damage may explain why these cells did not remain in G2 but instead re-initiated DNA replication (Flatt et al., 2000). Therefore, based on our results, pocket proteins are probably not required for an efficient and complete G2 block in the presence of an active p53 pathway, but they might be essential to induce a prompt cell cycle exit. Their key role for G1 exit has been demonstrated for MEFs (Sage et al., 2000). This conclusion is supported by recent results showing that functional inactivation of pRb in NHFs induced apoptosis under the conditions in which wild-type fibroblasts undergo premature senescence (Lodygin et al., 2002).
In summary, the mechanism described in this work may be important to ensure permanent G2 cell cycle exit in primary human cells, favouring their survival while preventing propagation of potentially tumorigenic cells containing mutated or rearranged DNA.
Materials and methods
Cell culture
The NHFs used for this study included early passage fibroblasts isolated from neonatal human foreskin as described previously (Dazard, 2000) and IMR-90 (purchased from ATTC, population doubling (P.D. ∼26). They were cultured in DMEM or MEM/F12 supplemented with 10% FCS. IMR-90 cells, at P.D. ∼30, were infected by retroviral transfer of the HPV-16 E6 and E7 oncogenes in the PLXSN vector, provided generously by Dr J.Shay (University of Texas Southwestern Medical Center, Dallas, TX) and Dr D.Galloway (Fred Hutchinson Cancer Research Center, Seattle, WA). The cells were selected for resistance to G418 and were passaged according to the same protocol used for wild-type NHFs (Dulić et al., 1998). Wild-type MEFs were a generous gift from Dr U.Hibner (IGM, Montpellier, France).
Cell synchronization and drug treatment
The cell were synchronized by two protocols. NHFs (control and E6 cells) were pre-synchronized in G0 by contact inhibition and then released into the cell cycle by dilution. Cell cycle arrest at the G1/S boundary was accomplished by the addition of 2 mM hydroxyurea (HU) for 12 h. HU was removed by extensive washing and the cells were harvested at different times after release from the block. The synchrony of the population was determined by flow cytometric analysis (FACS) after staining of nuclear DNA with propidium iodide. For E7-expressing fibroblasts, exhibiting inefficient G0 arrest, the cells were synchronized at the G1/S boundary by the addition of aphidicolin (2.5 µM per 24 h). The drugs were added 3 h after release from the G1/S block at the previously established concentrations: 2 µg/ml ICRF-193 and 10 µg/ml bleomycin. To determine the capacity of the cells to enter the cell cycle once arrested in G2, the drugs were removed by extensive washing and BrdU incorporation was monitored at different times.
Microscopy
The conditions for cell fixation, indirect immunofluorescence and staining of the nuclei by Hoechst 33258 (Sigma) were the same as described previously (Dulić et al., 1998). SA-β-gal staining was performed according to the published protocol (Dimri et al., 1995). Video-microscopy experiments (Hamamatsu software) were carried out at the ‘Integrated Image Facility’ (CRBM) directed by Dr P.Travo.
Immunoprecipitations, immunoblot analysis and antibodies
Preparation of whole cell lysates from frozen cell pellets, conditions for immunoprecipitation, histone H1 kinase assays and immunoblotting have been described previously (Dulić et al., 1994). For p21 depletion, total cell extracts (100–150 µg) were incubated with saturating amounts of p21-specific antibodies, whereas mock samples were incubated with protein A Sepharose beads only. The resulting supernatants were analysed by immunoblotting and/or used for further immunoprecipitation with cyclin-specific antibodies as described previously (Dulić et al., 1998). In some cases, the same immunoprecipitates were used both for determination of histone H1 kinase activity and for immunoblot detection of various components by a controlled partial transfer of SDS–PAGE gels. The resulting membrane was used for immunoblot analysis, whereas the gel was stained, dried and the histone H1 bands excised and counted as described previously (Dulić et al., 1994).
Most of the primary antibodies used in this study were the same as described in earlier publications (Dulić et al., 1998, 2000). In addition, we have used polyclonal anti-cyclin B1 (sc-752), polyclonal anti-Cdk2 (sc-163), polyclonal anti-p130 (sc-317), polyclonal anti-p107 (sc-318) and monoclonal anti-p53 (DO-1, sc-126), all from Santa-Cruz Biotechnology, and monoclonal anti-phospho histone H3 (6G3) and polyclonal antibody anti-P-T161-Cdk1 from Cell Signalling Technology. Secondary antibodies for western blot analysis were anti-mouse and anti-rabbit IgG–HRP conjugates (Promega) and protein AG coupled to HRP (Pierce). Proteins were visualized by an enhanced chemiluminescence detection system according to the manufacturer (Amersham).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Dr Marcel Dorée for invaluable comments and critical suggestions and Dr Pierre Travo (CRBM) for his enthusiastic help with the time-lapse microscopy. We are also grateful to Dr Ursula Hibner for MEFs, Drs Jerry Shay and Denise Galloway for reagents used in retroviral transfections, and Dr A.M.Creighton for the generous gift of ICRF-193. F.B. is recipient of a fellowship from the Ministère de l’Education Nationale et de la Recherche (MENR), and V.G. is recipient of a fellowship from the Association de la recherche sur le cancer (ARC). This work was supported by grants from La Ligue nationale contre le cancer (to V.D., équipe labelisée), ARC (to J.P.) and MENR (to J. P.).
References
- Abraham R.T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev., 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- Agarwal M.L., Agarwal,A., Taylor,W.R. and Stark,G.R. (1995) p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl Acad. Sci. USA, 92, 8493–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen P.R., Lacroix,F.B., Lohez,O.D. and Margolis,R.L. (2001a) Neither p21(WAF1) nor 14-3-3σ prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res., 61, 7660–7668. [PubMed] [Google Scholar]
- Andreassen P.R., Lohez,O.D., Lacroix,F.B. and Margolis,R.L. (2001b) Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell, 12, 1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel F., Lohez,O.D., Lacroix,F.B. and Margolis,R.L. (2002) Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl Acad. Sci. USA, 99, 9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux,A., Lengauer,C., Waldman,T., Zhou,S., Brown,J.P., Sedivy,J.M., Kinzler,K.W. and Vogelstein,B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science, 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Dazard J.-E. (2000) Rôle régulateur des protéines p53 et Mdm2 au cours de la différenciation terminale kératinocytaire. PhD thesis, Université de Montpellier I, France.
- Deming P.B., Cistulli,C.A., Zhao,H., Graves,P.R., Piwnica-Worms,H., Paules,R.S., Downes,C.S. and Kaufmann,W.K. (2001) The human decatenation checkpoint. Proc. Natl Acad. Sci. USA, 98, 12044–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P. et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes C.S., Clarke,D.J., Mullinger,A.M., Gimenez-Abian,J.F., Creighton,A.M. and Johnson,R.T. (1994) A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature, 372, 467–470. [DOI] [PubMed] [Google Scholar]
- Dulić V., Kaufmann,W.K., Wilson,S.J., Tlsty,T.D., Lees,E., Harper,J.W., Elledge,S.J. and Reed,S.I. (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell, 76, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Dulić V., Stein,G.H., Far,D.F. and Reed,S.I. (1998) Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol. Cell. Biol., 18, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulić V., Beney,G.E., Frebourg,G., Drullinger,L.F. and Stein,G.H. (2000) Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol. Cell. Biol., 20, 6741–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatt P.M., Tang,L.J., Scatena,C.D., Szak,S.T. and Pietenpol,J.A. (2000) p53 regulation of G2 checkpoint is retinoblastoma protein dependent. Mol. Cell. Biol., 20, 4210–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuno N., den Elzen,N. and Pines,J. (1999) Human cyclin A is required for mitosis until mid prophase. J. Cell Biol., 147, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottifredi V., Karni-Schmidt,O., Shieh,S.S. and Prives,C. (2001) p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol. Cell. Biol., 21, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grana X., Garriga,J. and Mayol,X. (1998) Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene, 17, 3365–3383. [DOI] [PubMed] [Google Scholar]
- Hu B., Mitra,J., van den Heuvel,S. and Enders,G.H. (2001) S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant-negative mutant in human cells. Mol. Cell. Biol., 21, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida R., Sato,M., Narita,T., Utsumi,K.R., Nishimoto,T., Morita,T., Nagata,H. and Andoh,T. (1994) Inhibition of DNA topoisomerase II by ICRF-193 induces polyploidization by uncoupling chromosome dynamics from other cell cycle events. J. Cell Biol., 126, 1341–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann W.K. and Kies,P.E. (1998) DNA signals for G2 checkpoint response in diploid human fibroblasts. Mutat. Res., 400, 153–167. [DOI] [PubMed] [Google Scholar]
- Lodygin D., Menssen,A. and Hermeking,H. (2002) Induction of the Cdk inhibitor p21 by LY83583 inhibits tumor cell proliferation in a p53-independent manner. J. Clin. Invest., 110, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe S.W., Schmitt,E.M., Smith,S.W., Osborne,B.A. and Jacks,T. (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature, 362, 847–849. [DOI] [PubMed] [Google Scholar]
- Mantovani F. and Banks,L. (2001) The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene, 20, 7874–7887. [DOI] [PubMed] [Google Scholar]
- Munger K., Basile,J.R., Duensing,S., Eichten,A., Gonzalez,S.L., Grace,M. and Zacny,V.L. (2001) Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene, 20, 7888–7898. [DOI] [PubMed] [Google Scholar]
- Ohi R. and Gould,K.L. (1999) Regulating the onset of mitosis. Curr. Opin. Cell Biol., 11, 267–273. [DOI] [PubMed] [Google Scholar]
- Pagano M., Pepperkok,R., Verde,F., Ansorge,W. and Draetta,G. (1992) Cyclin A is required at two points in the human cell cycle. EMBO J., 11, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passalaris T.M., Benanti,J.A., Gewin,L., Kiyono,T. and Galloway,D.A. (1999) The G2 checkpoint is maintained by redundant pathways. Mol. Cell. Biol., 19, 5872–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.Y., Graves,P.R., Thoma,R.S., Wu,Z., Shaw,A.S. and Piwnica-Worms,H. (1997) Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science, 277, 1501–1505. [DOI] [PubMed] [Google Scholar]
- Ren B., Cam,H., Takahashi,Y., Volkert,T., Terragni,J., Young,R.A. and Dynlacht,B.D. (2002) E2F integrates cell cycle progression with DNA repair, replication and G2/M checkpoints. Genes Dev., 16, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J., Mulligan,G.J., Attardi,L.D., Miller,A., Chen,S., Williams,B., Theodorou,E. and Jacks,T. (2000) Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev., 14, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M., Huibregtse,J.M., Vierstra,R.D. and Howley,P.M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell, 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Smits V.A., Klompmaker,R., Vallenius,T., Rijksen,G., Makela,T.P. and Medema,R.H. (2000) p21 inhibits Thr161 phosphorylation of cdc2 to enforce the G2 DNA damage checkpoint. J. Biol. Chem., 275, 30638–30643. [DOI] [PubMed] [Google Scholar]
- Stewart Z.A., Leach,S.D. and Pietenpol,J.A. (1999) p21Waf1/Cip1 inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol. Cell. Biol., 19, 205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor W.R. and Stark,G.R. (2001) Regulation of the G2/M transition by p53. Oncogene, 20, 1803–1815. [DOI] [PubMed] [Google Scholar]
- Taylor W.R., Schonthal,A.H., Galante,J. and Stark,G.R. (2001) p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J. Biol. Chem., 276, 1998–2006. [DOI] [PubMed] [Google Scholar]
- Winters Z.E., Ongkeko,W.M., Harris,A.L. and Norbury,C.J. (1998) p53 regulates Cdc2 independently of inhibitory phosphorylation to reinforce radiation-induced G2 arrest in human cells. Oncogene, 17, 673–684. [DOI] [PubMed] [Google Scholar]
- Yang J., Bardes,E.S., Moore,J.D., Brennan,J., Powers,M.A. and Kornbluth,S. (1998) Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev., 12, 2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]