

Abstract
Muscarinic acetylcholine receptors consist of five distinct subtypes and have been important targets for drug development. In the periphery, muscarinic acetylcholine receptors mediate cholinergic signals to autonomic organs, but specific physiological functions of each subtype remain poorly elucidated. Here, we have constructed and analyzed mutant mice lacking the M3 receptor and have demonstrated that this subtype plays key roles in salivary secretion, pupillary constriction, and bladder detrusor contractions. However, M3-mediated signals in digestive and reproductive organs are dispensable, likely because of redundant mechanisms through other muscarinic acetylcholine receptor subtypes or other mediators. In addition, we have found prominent urinary retention only in the male, which indicates a considerable sex difference in the micturition mechanism. Accordingly, this mutant mouse should provide a useful animal model for investigation of human diseases that are affected in the peripheral cholinergic functions.
Muscarinic acetylcholine receptors (mAChRs) belong to a group of seven transmembrane-spanning receptors and are distributed widely in both central and peripheral nervous systems. The M3 type of affinity has been identified through ligand-binding studies (1) as that found in the salivary gland and can be distinguished from that found in the cerebral cortex (M1) or in the myocardium (M2). Molecular cloning studies have revealed five distinct genes for mAChRs in rats and humans, and it is now generally accepted that five classes of affinity (M1–M5) are attributable to these gene products, respectively (2). Previously, we mapped five mouse genes for mAChR (Chrm1, Chrm2, Chrm3, Chrm4, and Chrm5) on mouse chromosomes 19, 6, 13, 2, and 2, respectively (3). Among them, the M1, M3, and M5 receptors are usually coupled to the Gq/11 protein, which activates phospholipase C, whereas the M2 and M4 receptors are mainly coupled to the Gi/o protein, which inhibits adenylate cyclase activity.
The M3 subtype of mAChR is distributed widely in the peripheral autonomic organs, with the highest expression found in the exocrine glands and smooth muscle tissues. Whereas multiple subtypes of mAChRs coexist in the same tissue, it has been suggested through in vitro studies that the most abundant subtype is not necessarily predominant in exerting biological effects. In many smooth muscle tissues, for example, the M3 receptors have been demonstrated to play dominant roles in eliciting cholinergic contraction in vitro, despite the fact that the major acetylcholine-binding site shows the M2 type of affinity (4). However, further characterization of physiological roles of each mAChR subtype in vivo has been hampered by lack of subtype-specific ligands.
Determining the subtype-specific functions has been a matter of considerable interest because mAChRs are suitable targets for various therapeutic drugs (5). For example, an M1-selective antagonist, pirenzepine, has been used widely for treating peptic ulcer. Recently, several M3-selective antagonists were developed for use in airway obstructive diseases, urge incontinence, and irritable bowel syndrome. In this regard, knowledge of the phenotype caused by loss of the M3 receptor should be useful for evaluating these drugs in the respective organs. Although ligands for mAChRs active in the brain can be promising drugs for neurodegenerative disorders such as Alzheimer's disease, their clinical applications are often limited by their considerable side effects in the periphery. Accordingly, studies on the M3-mediated in vivo functions should help develop better cholinomimetic drugs with more subtype specificity and less undesirable side effects.
Recently, knockout mice for the M1, M2, and M4 receptors were reported (6–8). In these papers, the authors have characterized the roles of the respective subtypes in the central nervous system, but peripheral autonomic functions of these mutant mice appear intact, suggesting the primary importance of the M3 subtype in the peripheral cholinergic systems.
In this study, we generated Chrm3 null mutant mice and analyzed their phenotypes to determine the physiological role of this receptor subtype.
Materials and Methods
Construction of a Targeting Vector for Chrm3 Disruption.
The genomic cloning of Chrm1 and Chrm3 from a mouse 129/SvJ library was described previously (3). A targeting vector, pChrm3-N2 (Fig. 1A), was constructed from the inserts of the phage clones. The NcoI–XbaI (1.0 kb) and Sse8387I–NotI (9.0 kb) fragments were placed upstream and downstream of the phosphoglycerate kinase I promoter (PGK)-neo-bpA cassette (9), respectively. Finally, the PGK-DTA cassette (10) was inserted at the upstream end in the reverse orientation.
Figure 1.
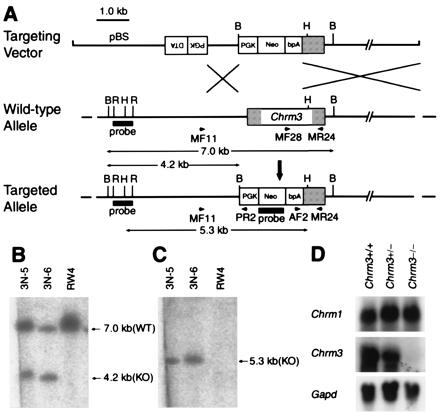
Generation of Chrm3-deficient mice. (A) Targeting strategy by homologous recombination in ES cells. Targeting vector pChrm3-N2 contained the neo gene and the diphtheria toxin α-subunit gene (DTA; ref. 10) driven by the phosphoglycerate kinase I promoter. Arrows MF11 and PR2 indicate the PCR primers used for homologous recombinant screening, whereas MF28, AF2, and MR24 for genotyping. BamHI (B), HindIII (H), and EcoRI (R) sites relevant to the identification of homologous recombinant ES cell clones are shown together with the expected sizes hybridizable to the Chrm3 and the neo probes. (B and C) Confirmation of the homologous recombination in ES cell clones by Southern hybridization. (B) Hybridization with the Chrm3 probe showing a 4.2-kb band specific to the targeted allele and a 7.0-kb band derived from the wild-type allele. (C) Hybridization with the neo probe showing a 5.3-kb band specific to the targeted allele. (D) Northern analysis showing mRNA levels of Chrm1 and Chrm3 in the brains of wild-type, heterozygous, and homozygous mice. Note that the Chrm3 mRNA in the heterozygous brain decreased to about half of the wild-type brain and is absent in the homozygous brain. The mRNA levels of Chrm1 were not different among the three genotypes. (Bottom) Signals hybridized with a Gapd probe used as an internal control.
Gene Targeting in Embryonic Stem (ES) Cells.
ES cells (2.0 × 107; RW4 clone; Genome Systems, St. Louis) were electroporated with the targeting vector linearized at the unique NotI site. G418-resistant homologous recombinant candidates were screened by PCR of 43 cycles (30 sec at 94°C, 30 sec at 55°C, and 60 sec at 72°C) after activation of AmpliTaq Gold (Perkin–Elmer) by heating 10 min at 95°C, by using primers MF11 (5′-AGC AGA ACC CTG AAC CAT GC-3′) and PR2 (5′-TAA AGC GCA TGC TCC AGA CT-3′) to amplify a 1.0-kb fragment. The PCR mixture consisted of 0.2 μM of each primer, 200 μM dNTPs, 1.5 mM MgCl2, and 0.5 units AmpliTaq Gold polymerase in PCR Buffer II in a final volume of 20 μl. Homologous recombination of the candidate clones were verified further by Southern hybridizations (Fig. 1 B and C). Chimeric mice were generated by injecting the ES cells into C57BL/6 blastocysts (11) and transferred to pseudopregnant MCH females (CLEA Japan, Tokyo). The lights in the animal room were turned on between 7:00 a.m. and 7:00 p.m. The mice were fed standard dry pellets, CA-1 (CLEA), and water ad libitum. To improve the growth of the homozygous pups, hydrated paste food was prepared by mixing the powder form of CA-1 (CLEA) with twice the weight of sterilized tap water and was given to the litters from the age of 2 weeks until the body weight reached about 20 g (males) or 19 g (females).
Genotyping.
Tail DNA was extracted by phenol and chloroform from proteinase K-digested samples. PCR amplifications for genotyping were performed in a single reaction mixture by using the following primers: MF28 (5′-AAG GCA CGA AAC GGT CAT CT-3′), AF2 (5′-GGG AAG ACA ATA GCA GGC AT-3′), and MR24 (5′-GCA AAC CTC TTA GCC AGC GT-3′). Other PCR conditions were the same as described above.
Northern Analysis.
Total RNA was extracted from the whole brain tissue of 3-month old male mice with ISOGEN (NipponGene, Toyama, Japan) according to the manufacturer's instructions. Northern hybridization analysis was performed according to a standard protocol (12) with minor modifications by using a positively charged nylon membrane, Hybond-N+ (Amersham Pharmacia Biotech). To prepare probes, DNA fragments corresponding to parts of coding regions of Chrm1, Chrm3, and the glyceraldehyde-3-phosphate dehydrogenase gene (Gapd) were amplified by PCR from cloned genomic DNA fragments of a 129/SvJ mouse (Chrm1 and Chrm3) or from a genomic DNA of a C57BL/6 mouse (Gapd). The primers used were: for Chrm1, Chrm1F (5′-GCA GCA GCT CAG AGA GGT CAC AG-3′) and Chrm1R (5′-GAT GAA GGC CAG CAG GAT GG-3′); for Chrm3, Chrm3F (5′-AAG GCA CGA AAC GGT CAT CT-3′) and Chrm3R (5′-GCA AAC CTC TTA GCC AGC GT-3′); and for Gapd, GapdF (5′-GCG TCC TGC ACC AAC TG-3′) and GapdR (5′-ATG GTC CTT TAC TCG AAG TG-3′). The PCR products for Chrm1 and Chrm3 were used directly as templates to synthesize 32P-labeled probes. The PCR product for Gapd was subcloned in the pGEM-T Easy vector (Promega), its identity confirmed by a restriction analysis, and it was used as a template to synthesize probes. Hybridizations were performed by using ExpressHyb Hybridization Solution (CLONTECH) according to the manufacturer's instructions.
Salivary Secretion Induced by Cholinergic and Adrenergic Agonists.
Total saliva was collected from 7- to 8-month old male mice. Animals were fasted for 5 to 7 hours before saliva collections that were performed between 3:00 and 6:00 p.m. After general anesthesia by i.p. injection of pentobarbital (50 mg/kg), pilocarpine (1.0 mg/kg) or isoproterenol (0.3 mg/kg) dissolved in saline was injected s.c. (10 ml/kg). Each saliva sample was collected for 30 min from the oral cavity by micropipette, and its volume was measured by using a 200-μl micropipette.
Histological Analyses.
All organs were fixed in 10% formalin, embedded in paraffin, and sectioned at 4 μm, except that the salivary gland was sectioned at 2 μm. Sections were stained with hematoxylin and eosin and examined under a light microscope.
In Vitro Responsiveness of Ileal and Urinary Bladder Smooth Muscles.
Adult mice (3–8 months old) were killed by cervical dislocation. The urinary bladder was prepared free of serosal connective tissue and cut into four longitudinal unfolded sections. The ileum was removed, and longitudinal muscles were isolated from the segments of the ileum (5 mm in length). The preparations were placed in 5-ml organ baths containing modified Krebs–Henseleit solution maintained at 32°C, continuously aerated with 95% O2 and 5% CO2, and connected to isometric transducers with sutures.
Mechanical responses were recorded isometrically by a multichannel polygraph. The tissue was equilibrated for at least 60 min with initial tension of 0.5 g, and then contractions were induced by adding 50 mM KCl twice. The second contractile response to KCl was taken as a reference. The concentration–contraction curves for carbachol were obtained by cumulative addition of carbachol to the organ bath. The pKB value, as an index of potency, was determined for each individual curve by the equation KB = B/(concentration ratio −1), where concentration ratio is the ratio of EC50 values with or without the antagonist, and B is the concentration of the antagonist (M).
Blood Chemistry.
Blood samples were taken intracardially from anesthetized animals (5–7 months old) and analyzed with a FUJI DRI-CHEM system (Model 5500) at Fujimoto Biomedical Laboratories (Osaka, Japan).
Drugs.
The following drugs were used in the experiments: atropine sulfate (Sigma); carbachol (carbamylcholine chloride; Sigma); (±)-isoproterenol hydrochloride (Sigma); methoctramine tetrahydrochloride (Research Biochemicals International, Natick, MA); pentobarbital sodium (Nembutal; Abbott); pilocarpine hydrochloride (Sigma); and pirenzepine dihydrochloride (Sigma).
Results
Chrm3 Gene Targeting in Mice.
As shown in Fig. 1A, a 1.5-kb XbaI-Sse8387I fragment of the mouse Chrm3 gene including the translation initiation codon was deleted and replaced with a phosphoglycerate kinase I promoter-neo-bpA cassette (9), by homologous recombination in ES cells of the 129/SvJ origin. Of 36 G418-resistant ES cell clones, two independent clones were identified as homologous recombinant candidates by PCR and verified for the homologous recombination by Southern hybridizations (Fig. 1 B and C). On injection into C57BL/6 blastocysts, one of the recombinant ES clones (3N-5) contributed to chimeric males, which transmitted the targeted allele to the germ line. A mutant mouse line was established by backcrossing with C57BL/6, i.e., in a mixed background between 129/SvJ and C57BL/6.
Growth Retardation of the Homozygous Mutant Mice.
The appearance of neonates derived from heterozygous intercrosses was normal. Then we noticed that, at around 3 weeks of age, the body size of some pups was smaller than that of their littermates. All these small pups were homozygous for the mutant allele and were born at the Mendelian ratio (+/+: +/−: −/− = 46: 86: 48). The weight gain of the homozygous pups was slow for the next few weeks, and their average body weight was only half that of their littermates at 4–5 weeks of age (Fig. 2 A and B). Most homozygous pups (40/48), however, survived this critical period. Thereafter, the time courses of growth recovery were different between the sexes. Namely, the homozygous males caught up with their littermates in body weight, but homozygous females remained smaller and were about 80% of the wild-type females at 15 weeks of age. To determine the cause of the growth retardation, we switched the diet from dry pellets to hydrated paste. With this measure, the growth of the homozygotes of both sexes improved significantly, as shown in Fig. 2 A and B. We found no apparent behavioral abnormalities of the homozygous mutants without specific experimentation. Effects of the M3 mutation on the central nervous system functions remain to be investigated systematically.
Figure 2.
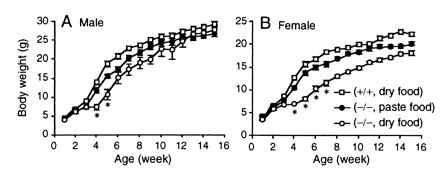
Growth curves of male (A) and female (B) mice. The value of each point is the average body weight (±SEM) of the wild-type and homozygous mutant mice (n = 5 to 7) monitored for 15 weeks. When fed a standard pellet diet, the growth of the homozygous mice (○) was retarded significantly, compared with that of the wild-type mice (□). Asterisks (*) indicate body weights 70% or less than those of wild-type mice. In both sexes, the weight differences became noticeable around 3 weeks of age and were most prominent at 4–5 weeks. When fed with hydrated paste diet from 2 weeks of age, the growth of the homozygous pups (●) improved significantly.
Northern Analysis of the M1 and M3 Receptor Gene Expression.
To confirm the loss of the M3 receptor, we determined the amounts of Chrm3 mRNA by Northern analysis in the total RNA extracted from the whole brain. As shown in Fig. 1D, Chrm3 mRNA was absent in the homozygous mutant tissue, and the amount in the heterozygous tissue was reduced to about half that in the wild type. When the same membrane was rehybridized with a Chrm1 probe, the mRNA was detected at similar levels among the three genotypes.
Preserved Reproductive Abilities in the Homozygous Mice.
The M3 receptors are expressed in the genital organs of both sexes. To evaluate the functional consequence of the M3 receptor loss in reproduction, we set up two mating pairs between homozygous mice (3-month old males and 4-month old females). In both pairs, females delivered pups, and their growth (nursed by the homozygous dams) was indistinguishable from that of pups born from the heterozygous intercrosses (data not shown).
Dilated Ocular Pupils and Their Responses to Cholinergic Agents.
As expected from the established role of the mAChR in pupillary constriction, all homozygotes had larger pupils. When observed in a bright room (≈1,000 lux), the pupil diameter of the homozygotes (3–4 months old) was 1.58 ± 0.06 mm (n = 12), whereas those of their heterozygous and wild-type littermates were 0.34 ± 0.01 mm (n = 14) and 0.30 ± 0.06 mm (n = 4), respectively (mean ± SEM), and there was no sex difference. We next photographed the eyes of an anesthetized heterozygote and homozygote (Fig. 3 A and B, respectively) under a brighter light (>20,000 lux) and found that the difference in pupil size was still apparent. However, the strongly illuminated pupils of the homozygote were slightly smaller (1.2 mm, measured from Fig. 3B) than those observed in the bright room, demonstrating a weak light reflex. When pilocarpine (a nonselective muscarinic agonist) solution was instilled, the pupil size changed very little in both mice (data not shown). In contrast, atropine instillation caused further dilatation and completed mydriasis in both mutants (Fig. 3 C and D). We found no other abnormalities, such as eyelid ptosis, depression of the eyeball, or redness of the conjunctiva, suggesting that sympathetic tone and basal lacrimal secretions were not affected significantly (data not shown).
Figure 3.
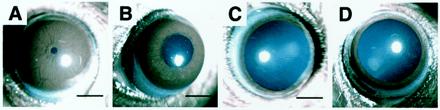
Appearance of pupils of the heterozygous (A and C) and homozygous (B and D) mutant mice (4-month old female littermates) on instillation of an antagonist (atropine) under bright illumination. (A and B) Before instillation. The pupils of the homozygotes (B) are larger than those of the heterozygous mouse (A). (C and D) Thirty minutes after an instillation of 1% atropine. Instillation of atropine caused full dilatation of the pupils not only in the heterozygous (C) but also in the homozygous mouse (D). Bars = 1 mm.
Impaired Salivary Secretion to Cholinergic Stimuli.
Because mAChRs mediate parasympathetic signals that stimulate serous salivary secretion (13), we evaluated a salivary gland function by measuring salivary flow rates stimulated by s.c. injection of pilocarpine (1.0 mg/kg) in anesthetized mice. Notably, the salivary response was reduced in the mutant mice, reflecting the gene dosage, to nearly undetectable levels in the homozygotes (Fig. 4 A and B). In contrast, isoproterenol (β-adrenergic agonist)-induced salivations were preserved in the mutant mice of both genotypes (Fig. 4 C and D). On histological examinations, the sections of the parotid, submandibular, and sublingual glands stained with hematoxylin and eosin appeared normal and showed no sign of atrophy or inflammation (data not shown).
Figure 4.
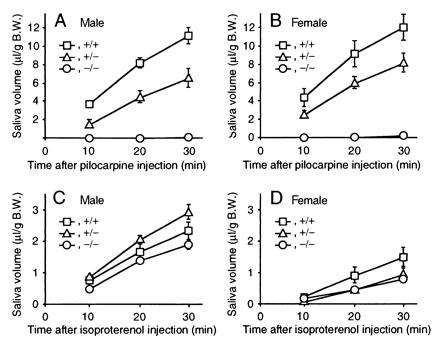
Stimulated salivary responses. (A) Response in the male mice on stimulation with pilocarpine. (B) Response in female mice on stimulation with pilocarpine. (C) Response in male mice on stimulation with isoproterenol. (D) Response in the female mice on stimulation with isoproterenol. Time courses of cumulative salivary volume (mean ± SEM) were shown that were determined at 10, 20, and 30 min after each stimulation. □, ▵, and ○ indicate the data of wild-type, heterozygous, and homozygous mice, respectively. Each symbol represents the data of four to five experiments.
Sex Difference in the Urinary Retention Phenotype.
In the adult homozygotes, most visceral organs such as the heart, lung, liver, and spleen appeared normal on gross and histological examinations (data not shown). Interestingly, however, all male homozygous mutants were found to have severely distended urinary bladders. The diameter of the homozygous male bladder (12.3 ± 1.3 mm, n = 6) was strikingly larger than that of the wild-type male bladder (5.4 ± 0.8 mm, n = 6). On histological examination, the bladder wall was thinner because of the severe distension (see a transverse section at the middle level of the bladder body shown in Fig. 5B) and the submucosal layer contained some focal lymphocyte infiltrations, whereas the urothelium appeared normal with no sign of dysplasia or hyperplasia (Inset). Consistent with the bladder phenotype, some homozygotes (3/10) showed mild hydronephrosis, i.e., the renal calyx was distended with some perivascular lymphocyte infiltrations in the adjacent renal medulla (Fig. 5D). Despite these apparent urinary flow disturbances, the urine of the homozygotes was clear, and their urinalysis data were normal (data not shown). The renal functions were not affected, as evidenced by the normal levels of blood urea nitrogen and creatinine (data not shown).
Figure 5.
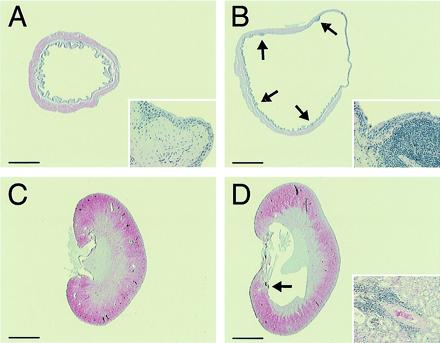
Histology of the bladder and kidney. (A and B) Male urinary bladders of wild-type (A) and homozygous (B) mice. The bladder of the homozygous mouse is distended with focal submucosal infiltrations of lymphocytes (arrows). Note that the smooth muscle layer is thinner, especially in the region of right upper quadrant, because of severe distension. Inset (×50 higher magnification) shows the normal epithelium with a submucosal lymphocyte infiltration. (C and D) Kidneys of wild-type (C) and homozygous (D) male mice. The renal calix of the homozygous mice is distended. Inset (×10) shows a representative lymphocyte infiltration in the perivascular region in the medulla (arrow). Bars = 1.5 mm (A and B) and 2.0 mm (C and D).
In contrast to this apparent urinary retention phenotype in the male mice, the bladder distension in the female homozygous mutants was mild. The bladder diameter of the homozygous female (5.7 ± 0.5 mm, n = 7) was significantly smaller than that of the homozygous male but still slightly larger than that of the wild-type female (4.2 ± 0.3 mm, n = 5). Consistent with this light urinary retention phenotype, the kidneys of the homozygous females appeared normal on gross and microscopic examinations (data not shown).
Impaired in Vitro Contractility of the Detrusor Smooth Muscle of Both Sexes.
To evaluate further the role of the M3 receptor in the bladder, we examined in vitro contractility of the detrusor muscles by carbachol stimulation. Whereas the homozygous muscles of both sexes responded normally to 50 mM KCl, their maximal responses to carbachol (a nonselective cholinergic agonist) were decreased to only 5% of those obtained in the wild-type muscles (Fig. 6 A and B). To estimate the subtype of mAChR that was responsible for the weak but significant residual contraction, we obtained the antagonistic profile for three muscarinic ligands that have different selectivities for respective mAChR subtypes. As a result, the contraction of the wild-type muscle was antagonized with the following rank order of pKB values: atropine (9.9) > pirenzepine (6.9) > methoctramine (5.9). In contrast, the rank order of potencies obtained with the homozygous muscle was: atropine (9.0) > methoctramine (7.4) > pirenzepine (6.4). Although the contraction of the homozygous detrusor muscles was too small to allow precise pharmacological evaluation, these data are most consistent with the M2-mediated residual contractions in both sexes (see Discussion).
Figure 6.
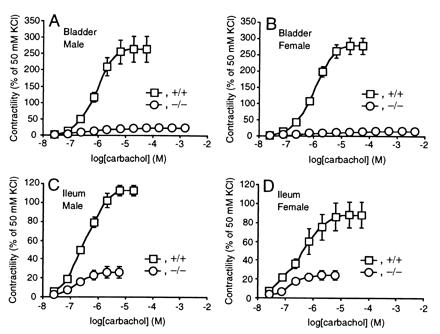
Reduced bladder (A and B) and ileal (C and D) smooth muscle contractions in response to carbachol in the homozygous mice. Responses to carbachol are shown (mean ± SEM) as percentages of those to 50 mM KCl in male (A and C) wild-type (n = 4) and homozygous (n = 4) mice and female (B and D) wild-type (n = 4) and homozygous (n = 4) mice. □ and ○ indicate the data of wild-type and homozygous mice, respectively.
Gastrointestinal Manifestations and Histology of the Intestines.
Through in vitro studies of the intestines, M3 has been considered to be a dominant subtype to mediate cholinergic signals both in the smooth muscle (14, 15) and mucosal epithelium (16). However, we found no apparent gastrointestinal complications such as diarrhea, constipation, or hemorrhage in any homozygotes. On necropsy, the length of the intestines was comparable among genotypes, and there was no considerable intestinal dilatation or intussusception. Histological examinations revealed no abnormalities in the hematoxylin and eosin-stained ileal or colonic sections (data not shown).
Impaired in Vitro Contractility of the Ileal Smooth Muscle.
To study further the role of the M3 receptor in the intestines, we examined in vitro contractility of the ileal longitudinal smooth muscle (Fig. 6 C and D). Its contractility to 50 mM KCl was comparable between the wild-type and homozygous tissues. However, the maximal contraction of the homozygous muscles to carbachol was decreased by 77 ± 5% in males and by 72 ± 6% in females (mean ± SEM, n = 4 for each sex per genotype, standardized by contractility to 50 mM KCl). The contraction of the wild-type muscle was antagonized with the following rank order of pKB values: atropine (9.4) > methoctramine (6.8) > pirenzepine (6.3), and with the homozygous mutant muscle; atropine (9.9) > methoctramine (8.2) > pirenzepine (6.6). These results suggest that the residual contraction was mediated mainly by the M2 receptor (see Discussion).
Discussion
Loss of the M3 receptor in mice has resulted in various overt phenotypes, as described above. These phenotypes are attributable to the impaired responses to cholinergic signals in the smooth muscle tissues and salivary glands. In contrast, mice lacking M1, M2, and M4 receptors (6–8) appeared normal, and their obvious phenotypes were revealed only after challenged by muscarinic agonists.
The homozygous pups were born at the Mendelian ratio and appeared normal until about 2 weeks of age, but they became significantly runted around weaning. Afterward, males slowly caught up with their littermates, whereas females remained smaller. It is worth noting that the growth of the inbred mice including the C57BL/6 strain delays temporarily at 3–4 weeks of age because of minor malnutrition on weaning (17). Therefore, it is most likely that transient growth retardation was caused by persistent malnutrition after weaning, because the homozygous mutants secreted very little saliva in response to pilocarpine even in the adults. A similar postweaning transient growth retardation was reported in the mice lacking neurturin receptor GFRα2 (18), which show developmental defects in the enteric and other parasympathetic nervous systems. Although the reason for the sex difference in the growth recovery remains to be investigated, it is conceivable that additional mechanisms such as defects in the endocrine system (19) are responsible.
Whereas salivary glands contain multiple subtypes of mAChRs (20), the markedly attenuated salivary response to pilocarpine in our homozygous mutant mice excludes any practical roles of other subtypes in salivary secretions. Interestingly, however, the oral cavities of the homozygotes were wet, and they were free of such complications as mucosal injury or dental caries that are found in severe xerostomia in the human (13). Accordingly, we propose that the basal level salivary secretion can be maintained by the sympathetic stimuli, whereas induction of additional salivation on eating requires the M3-mediated parasympathetic signals. Consistent with this mechanism, the salivary glands of the homozygotes had an intact histology and responded well to the β-adrenergic stimuli.
In the eye, parasympathetic stimulation of mAChRs causes contraction of the pupillary sphincter muscle. Whereas all subtypes of mAChRs are expressed in the muscle (21, 22), physiological contribution of each subtype has not been determined except for the M3 subtype (23). Our results provide genetic evidence for the key role of M3 in the normal pupillary constriction in mice. It should be noted, however, that a weak light reflex remained in the homozygous pupils, and that complete mydriasis was achieved only after an atropine instillation. These results provide further evidence that the other subtypes can play additional roles in miosis.
In the urinary bladder, we found that M3 receptors mediate 95% of the cholinergic contraction of the detrusor muscle in both sexes in vitro and that lack of the M3 receptor resulted in severe urinary retention in males in vivo. The former finding is consistent with an earlier report that suggests a functional predominance of the M3 subtype in the cholinergic signaling (24), and the latter reveals that parasympathetic comediators such as ATP (25) cannot substitute for acetylcholine in the micturition contraction in male mice. Although it is thought that cholinergic detrusor contraction is essential for the voiding process, the mild urinary phenotype in the homozygous females suggests that micturition in the female mice is rather regulated primarily by the relaxation of urethral muscles elicited by other mediators such as nitric oxide (26).
Similar chronic urinary retentions were reported in mice with a dysfunction of the nicotinic AChRs (nAChRs) in the autonomic ganglion caused by a targeted disruption of the α3 subunit gene (27) and in mice with disrupted β2 and β4 subunit genes (28). Although these mutant mice show additional urinary infections and stones and even develop such pathologic changes as hyperplasia and dysplasia of the bladder urothelium, it remains to be determined whether these additional phenotypes are caused directly by mutation(s) or by environmental complications. It is also worth noting that all autonomic input to the bladder should be blocked in nAChR mutant mice, whereas some other control mechanisms are expected to remain in M3 mutant mice, including the autonomic regulation by norepinephrine, endothelins, bradykinin, tachykinin peptides, ATP (25), and nitric oxide (26), as well as the regulation by ACh acting through the abundant M2 receptor. The lack of urinary stones and urothelial changes can be explained by the absence of urinary infection, because experimental urinary infections cause such complications (29, 30).
In the bladder (31) and intestinal smooth muscles (32–34), it has been demonstrated that cholinergic contractions in vitro are mediated predominantly by the M3 receptors. In contrast, stimulation of the most abundant subtype M2 (14, 15) has been suggested to exert minor indirect contraction by reversing relaxation induced by histamine or forskolin (33–35). In our observation, however, small but significant contractions remained in the homozygous muscles (bladder, 5%; ileum, 23–28%), even in the absence of such heterologous agents. To characterize these residual responses pharmacologically, we determined the pKB values for an M2-selective antagonist, methoctramine, and found that they were increased both in the bladder and ileal smooth muscles in the homozygotes (bladder, from 5.9 to 7.4; ileum, from 6.8 to 8.2). In addition, the low pKB values for pirenzepine in the homozygous muscles (bladder, 6.4; ileum, 6.6) ruled out any significant involvement of the M1 or M4 subtypes. Although further studies by using more selective agents may be needed for a more precise pharmacological evaluation, our results strongly suggest that the M2 receptors directly mediate the residual contraction of the M3 homozygous muscles. This interpretation is consistent with a recent report that homozygous M2 gene disruption in mice caused a slight but significant reduction in the smooth muscle contraction induced by carbachol (36).
It is worth noting that the homozygous mutant mice did not suffer from apparent gastrointestinal disorders, despite such in vitro defects in the ileal muscle contraction and the proposed role of the M3 receptor in controlling mucosal electrolyte transportations (16). Although the reason for this discrepancy remains to be investigated, it is conceivable that physiological functions of the digestive tract are controlled by a multitude of signaling mechanisms mediated by many neurotransmitters in the enteric nervous system (37).
Several lines of evidence suggested the importance of mAChRs in the reproductive organs of both sexes. For example, the vas deferens (38), prostate gland (39), and corpus cavernosum of penis (40) express mAChRs of the M3 profile. In addition, M3 receptors are responsible for uterine smooth muscle contraction (41). However, our result of crosses between homozygous mice suggests that M3 receptors are dispensable for the normal reproduction of the mouse.
Regarding possible similarities of the Chrm3 mutant phenotypes with human diseases, bilateral congenital mydriasis [Online Mendelian Inheritance in Man, OMIM, Johns Hopkins University, Baltimore, MD (http://www.ncbi.nlm.nih.gov/omim/), NCBI, National Library of Medicine, National Institutes of Health, no. 159420; ref. 42] is characterized by a similar ocular manifestation to that found in the homozygous mice. Although this disease is accompanied by growth retardation occasionally, all reported cases were in females and were without salivary or urinary symptoms. Another autosomal recessive disease, megacystis-microcolon-intestinal hypoperistalsis syndrome (OMIM no. 249210; ref. 43) shows a severe congenital urinary retention together with severe intestinal symptoms. However, no mydriasis has been reported in the disease, and the intestinal appearance of the homozygous mice was normal. Accordingly, neither of these human diseases appears to be caused by null mutations of the M3 mAChR gene (CHRM3).
In conclusion, we have constructed and analyzed Chrm3 null mutant mice and demonstrated that M3 receptors play key physiological roles in salivary secretion, pupillary constriction, and bladder detrusor contractions, whereas the role of M3 receptors in other organs are dispensable likely because of redundant mechanisms through other mAChR subtypes or other mediators. Mice lacking the M3 receptor should provide a useful animal model to investigate human diseases, such as glaucoma, dry eyes, and neurogenic bladder, which involve altered peripheral cholinergic functions.
Acknowledgments
We thank K. Noguchi, M. Kobayashi, M. Miyaji, and A. Sato for valuable discussion and T. Tamai, T. Ishikawa, and K. Takaku for technical advice. We also thank I. Ishii, A. Matsunaga, and A. Yokoi for blastocyst injections, Y. Araki, S. Kobayashi, and N. Matsubara for technical assistance, and S. Ishikawa and his staff for animal care. We thank T. Manabe for comments on the manuscript. This work was supported by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Science, Sports and Culture (11670083), Japan.
Abbreviations
- mAChRs
muscarinic acetylcholine receptors
- ES
embryonic stem
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mitchelson F. Pharmacol Ther. 1988;37:357–423. doi: 10.1016/0163-7258(88)90005-8. [DOI] [PubMed] [Google Scholar]
- 2.Caulfield M P, Birdsall N J M. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 3.Matsui M, Araki Y, Karasawa H, Matsubara N, Taketo M M, Seldin M F. Genes Genet Syst. 1999;74:15–21. doi: 10.1266/ggs.74.15. [DOI] [PubMed] [Google Scholar]
- 4.Ehlert F J, Sawyer G W, Esqueda E E. Life Sci. 1999;64:387–394. doi: 10.1016/s0024-3205(98)00584-0. [DOI] [PubMed] [Google Scholar]
- 5.Eglen R M, Choppin A, Dillon M P, Hegde S. Curr Opin Chem Biol. 1999;3:426–432. doi: 10.1016/S1367-5931(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton S E, Loose M D, Qi M, Levey A I, Hille B, McKnight G S, Idzerda R L, Nathanson N M. Proc Natl Acad Sci USA. 1997;94:13311–13316. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H, Wess J. Proc Natl Acad Sci USA. 1999;96:1692–1697. doi: 10.1073/pnas.96.4.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Proc Natl Acad Sci USA. 1999;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soriano P, Montgomery C, Geske R, Bradley A. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 10.Yagi T, Ikawa Y, Yoshida K, Shigetani Y, Takeda N, Mabuchi I, Yamamoto T, Aizawa S. Proc Natl Acad Sci USA. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Proc Natl Acad Sci USA. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown T, Mackey K. Curr. Protocols Mol. Biol., Unit 4.9.1. New York: Wiley; 1997. [Google Scholar]
- 13.Herrera J L, Lyons M F, 2d, Johnson L F. J Clin Gastroenterol. 1988;10:569–578. doi: 10.1097/00004836-198810000-00019. [DOI] [PubMed] [Google Scholar]
- 14.Giraldo E, Vigano M A, Hammer R, Ladinsky H. Mol Pharmacol. 1988;33:617–625. [PubMed] [Google Scholar]
- 15.Zhang L B, Horowitz B, Buxton I L. Mol Pharmacol. 1991;40:943–951. [PubMed] [Google Scholar]
- 16.O'Malley K E, Farrell C B, O'Boyle K M, Baird A W. Eur J Pharmacol. 1995;275:83–89. doi: 10.1016/0014-2999(94)00758-y. [DOI] [PubMed] [Google Scholar]
- 17.Laird A K, Howard A. Nature (London) 1967;213:786–788. doi: 10.1038/213786a0. [DOI] [PubMed] [Google Scholar]
- 18.Rossi J, Luukko K, Poteryaev D, Laurikainen A, Sun Y F, Laakso T, Eerikäinen S, Tuominen R, Lakso M, Rauvala H, et al. Neuron. 1999;22:243–252. doi: 10.1016/s0896-6273(00)81086-7. [DOI] [PubMed] [Google Scholar]
- 19.Pinter I, Makara G B, Acs Z. J Neuroendocrinol. 1996;8:935–939. doi: 10.1111/j.1365-2826.1996.tb00824.x. [DOI] [PubMed] [Google Scholar]
- 20.Levey A I. Life Sci. 1993;52:441–448. doi: 10.1016/0024-3205(93)90300-r. [DOI] [PubMed] [Google Scholar]
- 21.Gil D W, Krauss H A, Bogardus A M, WoldeMussie E. Invest Ophthalmol Visual Sci. 1997;38:1434–1442. [PubMed] [Google Scholar]
- 22.Ishizaka N, Noda M, Yokoyama S, Kawasaki K, Yamamoto M, Higashida H. Brain Res. 1998;787:344–347. doi: 10.1016/s0006-8993(97)01554-0. [DOI] [PubMed] [Google Scholar]
- 23.Hagan J J, van der Heijden B, Broekkamp C L. Naunyn-Schmiedebergs Arch Pharmacol. 1988;338:476–483. doi: 10.1007/BF00179317. [DOI] [PubMed] [Google Scholar]
- 24.Longhurst P A, Leggett R E, Briscoe J A. Br J Pharmacol. 1995;116:2279–2285. doi: 10.1111/j.1476-5381.1995.tb15065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Igawa Y, Mattiasson A, Andersson K E. Br J Pharmacol. 1993;109:473–479. doi: 10.1111/j.1476-5381.1993.tb13593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee J G, Wein A J, Levin R M. Pharmacology. 1994;48:250–259. doi: 10.1159/000139187. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis R A, Ou C N, Patrick J, Role L, De Biasi M, Beaudet A L. Proc Natl Acad Sci USA. 1999;96:5746–5751. doi: 10.1073/pnas.96.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W, Orr-Urtreger A, Nigro F, Gelber S, Sutcliffe C B, Armstrong D, Patrick J W, Role L W, Beaudet A L, De Biasi M. J Neurosci. 1999;19:9298–9305. doi: 10.1523/JNEUROSCI.19-21-09298.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson D E, Russell R G, Lockatell C V, Zulty J C, Warren J W, Mobley H L. Infect Immun. 1993;61:2748–2754. doi: 10.1128/iai.61.7.2748-2754.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis C P, Cohen M S, Gruber M B, Anderson M D, Warren M M. J Urol. 1984;132:1025–1031. doi: 10.1016/s0022-5347(17)49992-7. [DOI] [PubMed] [Google Scholar]
- 31.Wang P, Luthin G R, Ruggieri M R. J Pharmacol Exp Ther. 1995;273:959–966. [PMC free article] [PubMed] [Google Scholar]
- 32.Eglen R M, Harris G C. Br J Pharmacol. 1993;109:946–952. doi: 10.1111/j.1476-5381.1993.tb13712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas E A, Baker S A, Ehlert F J. Mol Pharmacol. 1993;44:102–110. [PubMed] [Google Scholar]
- 34.Thomas E A, Ehlert F J. J Pharmacol Exp Ther. 1994;271:1042–1050. [PubMed] [Google Scholar]
- 35.Hegde S S, Choppin A, Bonhaus D, Briaud S, Loeb M, Moy T M, Loury D, Eglen R M. Br J Pharmacol. 1997;120:1409–1418. doi: 10.1038/sj.bjp.0701048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stengel P W, Gomeza J, Wess J, Cohen M L. J Pharmacol Exp Ther. 2000;292:877–885. [PubMed] [Google Scholar]
- 37.Goyal R K, Hirano I. N Engl J Med. 1996;334:1106–1115. doi: 10.1056/NEJM199604253341707. [DOI] [PubMed] [Google Scholar]
- 38.Miranda H F, Duran E, Bustamante D, Paeile C, Pinardi G. Gen Pharmacol. 1994;25:1643–1647. doi: 10.1016/0306-3623(94)90366-2. [DOI] [PubMed] [Google Scholar]
- 39.Lau W A, Pennefather J N. Eur J Pharmacol. 1998;343:151–156. doi: 10.1016/s0014-2999(97)01535-5. [DOI] [PubMed] [Google Scholar]
- 40.Traish A M, Palmer M S, Goldstein I, Moreland R B. Receptor. 1995;5:159–176. [PubMed] [Google Scholar]
- 41.Choppin A, Stepan G J, Loury D N, Watson N, Eglen R M. Br J Pharmacol. 1999;127:1551–1558. doi: 10.1038/sj.bjp.0702696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caccamise W C, Townes P L. Am J Ophthalmol. 1976;81:515–517. doi: 10.1016/0002-9394(76)90312-3. [DOI] [PubMed] [Google Scholar]
- 43.Anneren G, Meurling S, Olsen L. Am J Med Genet. 1991;41:251–254. doi: 10.1002/ajmg.1320410224. [DOI] [PubMed] [Google Scholar]