

Abstract
Mononuclear cells from peripheral blood of thalassemic patients were treated with morpholino oligonucleotides antisense to aberrant splice sites in mutant β-globin precursor mRNAs (pre-mRNAs). The oligonucleotides restored correct splicing and translation of β-globin mRNA, increasing the hemoglobin (Hb) A synthesis in erythroid cells from patients with IVS2–654/βE, IVS2–745/IVS2–745, and IVS2–745/IVS2–1 genotypes. The maximal Hb A level for repaired IVS2–745 mutation was ≈30% of normal; Hb A was still detectable 9 days after a single treatment with oligonucleotide. Thus, expression of defective β-globin genes was repaired and significant level of Hb A was restored in a cell population that would be targeted in clinical applications of this approach.
One of the most common genetic diseases of mankind is β-thalassemia. It affects large populations in the Mediterranean basin, Middle East, South East Asia, and Africa. Approximately 80 million people are carriers of the thalassemia trait and the percentage of carriers worldwide is increasing. Concomitant increases in the number of patients, presently numbering several hundred thousands, are held down by high infant mortality in underdeveloped countries, by population screening, genetic counseling, and abortions; the growing need for clinical treatment is evident (1).
The disease is due to mutations causing defective β-globin gene expression and deficiency of β-globin and adult hemoglobin (Hb) A. Homozygotes or compound heterozygotes for severe defects are affected with thalassemia major or Cooley's anemia, lethal if untreated, and suffer pronounced anemia, bone deformities, and hepatomegaly and splenomegaly (2). Regular, lifelong transfusions combined with iron chelation constitute current treatment. Bone marrow transplantation, the only cure, is limited by the scarcity of suitable donors and facilities. Experimental protocols to stimulate synthesis of fetal hemoglobin by sodium butyrate or hydroxyurea (3–6) or β-globin gene repair or replacement (4, 7, 8) have not yet been fully tested at the clinical level. Clearly there is a need for alternative treatments to replace the costly and cumbersome transfusion regimen.
More than 100 thalassemic mutations causing defective β-globin gene expression and β-globin deficiency have been identified, but the ones causing aberrant splicing are among the most common (9). They are found in intron 1 (IVS1–5, IVS1–6, and IVS1–110) and intron 2 (IVS2–654, IVS2–705, and IVS2–745) of the β-globin gene (9–15). A mutation in codon 26 of the gene also results in activation of an aberrant splicing pathway and a mutated β-globin protein (βE) (16, 17). A common pathogenic feature of these mutations is activation of aberrant splice sites and modification of the splicing pathways, even though the correct splice sites remain potentially functional.
Aberrant splicing of several of the β-globin splicing mutants has been shown in cell free extracts (18), in transfected HeLa based cell lines (19–22), and in a transgenic mouse model (23). More importantly, in vitro and in transfected cell lines correct splicing was restored by blocking the aberrant splice sites with antisense oligonucleotides. Here we report that treatment of erythroid progenitors from peripheral blood of thalassemic patients carrying the IVS2–654 and −745 mutations with oligonucleotides antisense to aberrant splice sites in β-globin pre-mRNA efficiently restored correct splicing and production of significant amounts of Hb A. These results indicate that the oligonucleotides entered the erythroid progenitor cells, migrated to the nucleus, hybridized to the aberrant splice sites, and led to the formation of spliceosomes and subsequent splicing at the correct splice sites. In consequence, expression of a defective β-globin gene was repaired in a cell population that would constitute the target in clinical applications of this approach.
Materials and Methods
Mononuclear Cells.
Blood samples were from two Italian patients with β-thalassemia major, IVS2–745/IVS2–745 and IVS2–745/IVS2–1, and from a Thai patient with thalassemia intermedia, IVS2–654/βE. As controls, blood from an Italian subject heterozygous for the IVS2–745 mutation and from a normal subject also were used. Informed consent was obtained in accordance with Italian and Thai regulations.
Following the manufacturer's protocols, total mononuclear cells were isolated by Ficoll gradient (lymphocyte separation medium, ICN/Cappel, Aurora, OH), purified from the remaining red blood cells with ammonium chloride solution (StemCell, Vancouver, BC, Canada), and washed twice with Iscove's modified Dulbecco's medium, 2% fetal bovine serum. The cells were suspended at 2 × 106 cells per ml in the above medium, 30% fetal bovine serum, 1% BSA (the latter two from StemCell, Vancouver, BC.), 10−4 M 2-mercaptoethanol, 2 mM l-glutamine, 100 units/ml penicillin-streptomycin, 3 units/ml of recombinant human Epoetin α (Amgen, Thousand Oaks, CA), and 25 ng/ml recombinant mouse stem cell factor (R & D Systems, Minneapolis, MN), and incubated in 5% CO2 at 37°C. The above fresh medium (300 μl) containing double concentrations of Epoetin α and stem cell factor were added per milliliter on days 4 and 8 of culture; the same amount of old medium was subsequently replaced with 2× Epoetin α and 2× stem cell factor medium every 4 days.
To detect erythroid forming unit (CFU-E) and burst-forming unit erythroid (BFU-E) colonies in the mononuclear cell cultures the cells were plated at 2 × 105 cells per ml of methylcellulose containing culture medium (Stem Cell, Vancouver, BC). Hemoglobinized CFU-E and BFU-E colonies appeared at 8–18 days of culture.
Oligonucleotide Treatment.
The 18- or 25-mer morpholino oligonucleotides targeted to the 3′-cryptic splice site (3′-18, CAUUAUUGCCCUGAAAG, 3′-25, GTATCATTATTGCCCTGAAAGAAAG) and 18- or 25-mer morpholino oligonucleotides targeted to the aberrant 5′-splice sites in the IVS2–654 (654–18, GCUAUUACCUUAACCCAG, 654–25,TTGCTATTACCTTAACCCAGAAATT) or in the IVS2–745 mutants (745–25, CAGAATGGTACCTGGATTGTAGGTG) were used in the experiment. A morpholino oligonucleotide targeted to the α-globin gene (25 mer, CCCGCCGCTCACCTTGAAGTTGACC) was used as a negative control. Morpholino oligonucleotides (24) were prepared and purified at GeneTools (Corvallis, OR) and AVI Biopharma (Portland, OR).
Cellular delivery of the oligonucleotides was performed by passing 2 × 106 cells suspended at 37°C in 200 μl of culture media containing the desired concentration of morpholino oligonucleotide seven times through a syringe with a 25-gauge needle. After 1 h of incubation at 37°C, 800 μl of medium was added and incubation was continued at 37°C for the times indicated in the figure legends. Syringe loading of suspension cells was the equivalent of scrape loading for adherent cells (25) and allowed for penetration of concentrated oligonucleotides into the cells.
Isolation and Analysis of β-Globin mRNA.
The total cellular RNA was isolated with TRI-Reagent (Molecular Research Center, Cincinnati, OH) and (50–200 ng) analyzed by reverse transcription (RT)-PCR by using rTth DNA polymerase (Perkin–Elmer, Norwalk, CT) with 0.2 μCi of [α-32P]dATP per sample at up to 25 cycles. Assay of β-globin pre-mRNA was performed with forward and reverse primers spanning positions 21–43 of exon 2 (Fig. 1, primer b) and positions 119–142 of intron 2 (primer c) in β-globin pre-mRNA, respectively. Aberrant and correct splicing of human IVS2–745 β-globin pre-mRNA was detected with forward primer b and the reverse primer spanning positions 6–28 of exon 3 (primer d). Correction of human β-globin IVS2–654 pre-mRNA splicing in the compound heterozygote IVS2–654/βE was detected by using a forward allele-specific βA primer a (GCAAGGTGAACGTGGATGAAGTTGGTGTTG, positions 50–79 of β-globin exon 1) and the reverse primer d. The RT-PCR products were separated on a 7.5% nondenaturing polyacrylamide gel and detected by autoradiography. No product was detectable without the reverse transcription step.
Figure 1.
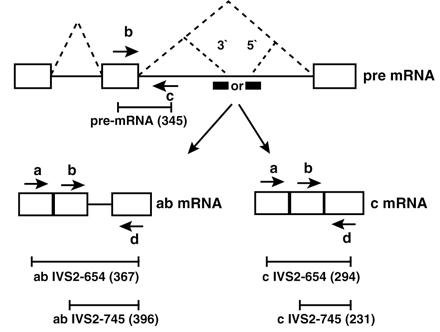
Correction of splicing of thalassemic human β-globin pre-mRNA by antisense oligonucleotides. Boxes, exons; solid lines, introns; dashed lines, correct and aberrant splicing pathways; the aberrant 5′-splice sites created by IVS2–654 and IVS2–745 mutations and the cryptic 3′-splice site activated upstream are indicated; heavy bars, oligonucleotide antisense to 3′- or 5′-splice sites; arrows, primers used in the RT-PCR reaction. The length (in nucleotides) of the appropriate RT-PCR products generated on aberrantly and correctly spliced mRNAs (ab and c mRNA, respectively) are indicated below each diagram.
Immunoblot Analysis of the Hemoglobins.
For hemoglobin analysis by cellulose acetate electrophoresis, 1.5 × 106 of the cultured mononuclear cells were washed twice with PBS, suspended in 20 μl of hemolysate reagent, and centrifuged at 14,000 rpm to remove the cell membranes. Approximately 3 μl of the supernatant was applied on Titan III-H cellulose acetate strips (76 × 60 mm) alongside with standard hemoglobins. Electrophoresis was performed with Supre-Heme buffer, pH 8.2 at 350 V for 35 min. The electrophoresis protocol and the materials were from Helena Laboratories (Beaumont, TX). Protein bands were visualized by staining the cellulose acetate strip with 0.5% Ponceau S and destaining with 5% acetic acid. Subsequently, the strip was blocked for 1 h in 5% solution of fat-free dry milk in PBS containing 0.1% Tween 20 and incubated with polyclonal affinity-purified chicken anti-human hemoglobin IgG as primary antibody and rabbit anti-chicken horseradish peroxidase-conjugated IgG as secondary antibody, both at 1,000-fold dilution in the blocking solution (Accurate, Westbury, NY). The blots were developed with an ECL detection system (Amersham, Piscataway, NJ).
Cytoimmunofluorescence.
The cultured cells were washed twice with PBS and loaded at 105 cells per well of printed microscope slides (Carlson Scientific, Peotone, IL). After air drying, the cells were fixed for 10 min in 1:1 methanol:acetone at −20°C. The slides were hydrated in PBS and incubated with mouse anti-human Hb A β8–12 mAb (Accurate, Westbury, NY), at 120 μg/ml for 1 h and with a secondary antibody, FITC anti-mouse donkey IgG1 diluted 200-fold for 1 h (Jackson ImmunoResearch Laboratories, West Grove, PA). Purified mouse IgG1 (Sigma, Saint Louis, MO) at 120 μg/ml served as the nonimmune control.
All autoradiograms were scanned using Adobe photoshop and figures were generated with Adobe ILLUSTRATOR. NIH IMAGE 1.61 software was used for quantitation.
Results
Experimental Approach.
A C to T mutation at nucleotide 654 (IVS2–654) and a C to G mutation at nucleotide 745 (IVS2–745) of β-globin intron 2 create aberrant 5′-splice sites at positions 652 and 745, respectively. These splice sites and a common cryptic 3′-splice site at nucleotide 579 are recognized by the splicing machinery, leading to inclusion of a fragment of the intron into the spliced mRNAs. The retained intron sequences generate a stop codon that prevents proper translation of the mRNA and causes a deficiency in β-globin and resultant β-thalassemia (10, 14) (Fig. 1). β-Globin pre-mRNA splicing in normal individuals and in thalassemic patients occurs in mononuclear erythroid progenitor and precursor cells and ceases when the cells mature to enucleated reticulocytes.
To determine the optimal conditions for antisense oligonucleotide treatments, a culture of mononuclear cells isolated from peripheral blood of a healthy volunteer was established, and the time course of β-globin pre-mRNA and mRNA expression was assayed by RT-PCR (see Materials and Methods). The initial lack of detectable levels of both these RNAs (Fig. 2, days 0–2) suggested that committed erythroid progenitors were absent in the cell preparation and that the pre-mRNA and spliced mRNA that appeared subsequently (Fig. 2) must have originated from a small number of stem cells or early progenitors present in the blood sample. These results indicated that the culture conditions allowed erythropoietic differentiation and transcription and processing of β-globin pre-mRNA, the target for antisense oligonucleotides. Erythroid differentiation was confirmed by the fact that hemoglobinized colonies appeared in cultures in methylcellulose containing medium (not shown). Note that the steady state level of β-globin mRNA is higher than that of its pre-mRNA, hence the detection of mRNA prior (Fig. 2, day 4) to pre-mRNA (Fig. 2, day 6).
Figure 2.
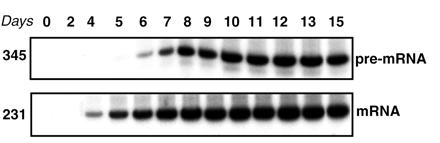
Time course of pre-mRNA and mRNA β-globin expression in cultured mononuclear cells from normal human blood assayed by RT-PCR of total RNA. (Upper) Expression of β-globin pre-mRNA; (Lower) expression of β-globin mRNA (see Materials and Methods). The numbers on the left indicate the size, in nucleotides, of the pre-mRNA and the correctly spliced β-globin mRNA.
Restoration of Correct Splicing of IVS2–654 β-Globin pre-mRNA.
On day 14 of culture, when a high level of β-globin pre-mRNA was expected (Fig. 2, days 13–15), the mononuclear cells from a thalassemic patient carrying the IVS2–654/βE mutations were treated with morpholino oligomers (24, 25) complementary to aberrant splice sites activated in IVS2–654 β-globin pre-mRNA. Morpholino analogs of oligonucleotides were selected because they are resistant to nucleases, do not allow degradation of RNA in the RNA-oligonucleotide hybrids by RNase H, and bind to targeted splice sites forming duplexes of high Tm (22).
Consistent with the β0 phenotype of IVS2–654 thalassemia (9), RT-PCR (primers a and d, Fig. 1) of total RNA from untreated cells (Fig. 3A, lane 1) detected only aberrantly spliced IVS2–654 β-globin mRNA and no correct β-globin mRNA. Primer a is specific for wild type β-globin, and therefore βE mRNA present in the sample (data not shown) was not detected. The 24-h treatment of the cells with 18- or 25-mer oligonucleotides targeted either to the cryptic 3′-splice site (3′−18 and −25, Fig. 3A, lanes 3–6) or the IVS2–654 5′-splice site (654–18 and −25, Fig. 3A, lanes 7–10) showed dose-dependent correction of aberrant splicing. The correction of β-globin splicing required a high concentration of the oligonucleotides, 45 μM, but was quite efficient in cultures treated with either 654–18 or 654–25 oligomers. Treatments with 3′-18 and −25 mers were less effective due to lower susceptibility of the cryptic 3′-splice site to the oligonucleotides (21).
Figure 3.
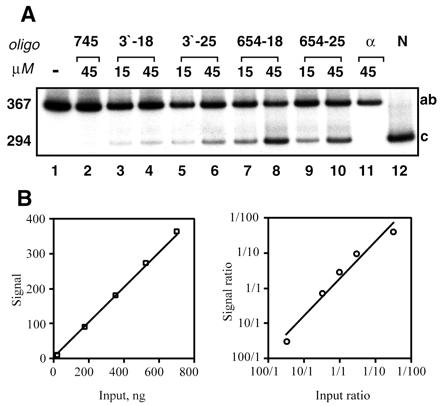
(A) Restoration of correct splicing of IVS2–654 β-globin pre-mRNA by antisense morpholino oligomers in cultured mononuclear cells from peripheral blood of IVS2–654/βE thalassemic patient (YC). RT-PCR analysis of total cellular RNA. On day 14 of culture, the cells were treated for 24 h with morpholino oligomers indicated at the top of the figure. Lanes 1, 2, and 11: control cells, untransfected, transfected with 745–25 mer, and with an α-globin-25mer, respectively. Note lack of production of correct β-globin mRNA. Lanes 3–6: cells transfected with oligonucleotides targeted to the 3′ cryptic splice site (3′−18 mer and -25 mer) at two different concentrations (15 and 45 μM). Lanes 7–10: cells transfected with oligonucleotides targeted to the aberrant 5′-splice site (654–18 mer and −25 mer), at 15 and 45 μM, each. Lane 12, β-globin mRNA from normal human blood. Due to the presence of correctly spliced mRNA derived from the βE gene (codon 26G→A), a 5′ βA codon 26 allele-specific forward primer was used (see Materials and Methods). The numbers on the left indicate the size, in nucleotides, of RT-PCR products representing the aberrantly (367) and correctly (294) spliced β-globin mRNAs. Unless otherwise noted similar designations are used in subsequent figures. (B) Linearity of RT-PCR assay. Increasing amounts (indicated on the Left panel) of total RNA from HeLa cells stably expressing correctly spliced β-globin mRNA were subjected to RT-PCR (20 cycles) and the products were analyzed and quantitated as described in Materials and Methods. Aberrantly spliced IVS2–654 RNA and correctly spliced β-globin RNA from HeLa cell lines were mixed in ratios indicated on the Right panel and analyzed as above. The results were averaged from three independent experiments.
The experiments illustrated in Fig. 3B show linearity and reproducibility of the RT-PCR protocol used. The amount of the PCR product was proportional to the amount of input RNA as were the relative amounts of PCR products generated from aberrantly and correctly spliced RNA (26). No product was detectable without the reverse transcription step (not shown). Quantitative analysis of the ratio of correct/aberrant RNAs showed that, at its best, repair of splicing yielded 35–50% of correct β-globin mRNA (Fig. 3, lanes 8 and 10). Control oligomers had no effect (Fig. 3, lanes 2 and 11), confirming the sequence specificity of correction; none of the oligomers showed any obvious cytotoxicity (not shown).
Hemoglobin A Synthesis in Oligonucleotide-Repaired IVS2–654/βE Erythroid Cells.
To test for hemoglobin synthesis, lysates of oligomer-treated cells separated by electrophoresis on cellulose acetate were probed with polyclonal anti-hemoglobin antibody (see Materials and Methods and Fig. 4). In untreated 8- and 10-day cultures of mononuclear cells (Fig. 4, lanes 1, 3, and 5), the only detectable species of hemoglobin were Hb E and Hb F. In a sample treated at day 8 and cultured for additional 3 days (lane 2), Hb A became detectable. The level of Hb A increased significantly upon treatment at day 10 (lane 4) and persisted for 5 days posttreatment (lane 6). Interestingly, in treated cultures the levels of Hb F decreased (see lanes 3 vs. 4 or 5 vs. 6), suggesting that restoration of expression of Hb A suppressed Hb F formation in repaired erythroid progenitor cells (lanes 2, 4, 6). However, the possibility that oligonucleotide treatment might have resulted in unspecific changes in γ-chain synthesis cannot be ruled out.
Figure 4.
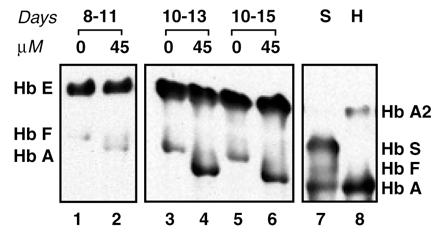
Immunoblot of hemolysate of mononuclear cells from patient YC probed with anti-human hemoglobin antibody. Concentrations of 654–18 mer antisense oligonucleotide are indicated above the lanes. Lanes 1–2, hemolysate from cells transfected on day 8 and harvested on day 11; lanes 3–6, hemolysate from cells transfected on day 10 and harvested on day 13 (lanes 3 and 4) and 15 (lanes 5 and 6); lane 7, hemoglobin standards (S); and lane 8, hemolysate of red blood cells from an IVS2–745 heterozygote subject (H).
Repair of Splicing and Hemoglobin A Synthesis in IVS2–745 Erythroid Cells.
A similar series of experiments was carried out with mononuclear cells from two patients with IVS2–745/IVS2–745 and IVS2–745/IVS2–1 thalassemias. In a 7-day culture from the homozygous patient, DD, aberrantly spliced mRNA (band of 396 nucleotides) and small amounts of correct β-globin mRNA (band 231) were detected in untreated cells or cells treated with the control oligomer (Fig. 5A, lanes 1 and 2, respectively). Treatment with morpholino oligomers targeted to the aberrant splice sites showed a dose-dependent restoration of correct β-globin splicing (Fig. 5A, lanes 3–8). In all samples, high levels of newly generated β-globin mRNA, exceeding those of aberrant RNA, were detected. This result suggests that, due to greater stability of correct β-globin mRNA, its steady state level is higher than that of its aberrant 745 counterpart. Even more efficient production of correct β-globin mRNA was accomplished by oligonucleotide treatment of 14 day cultures (Fig. 5B).
Figure 5.
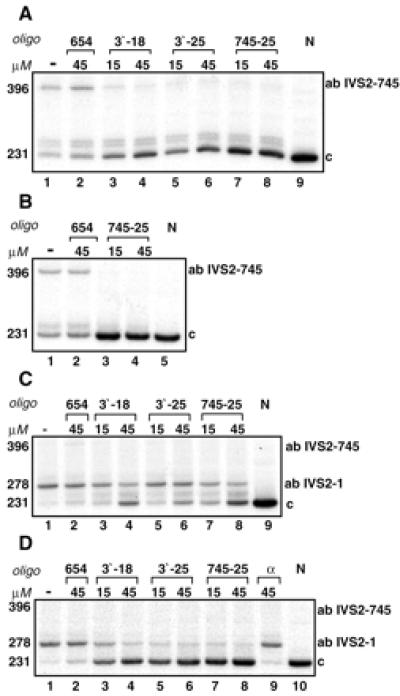
Correction of splicing of β-globin pre-mRNA in mononuclear cells from IVS2–745 homozygote (DD) and IVS2–745/IVS2–1 heterozygote (ZA) thalassemic patients. On days 7 (A) and 14 (B) of the of culture, the cells from patient DD were treated for 24 h with morpholino oligomers indicated above the lanes. N, RNA from normal blood (lanes 9 and 5 in A and B, respectively). Oligonucleotide targeted to the IVS2–654 aberrant 5′-splice site, uninvolved in splicing of IVS2–745 pre-mRNA served as negative control (lane 2 in A and B). (C and D) Analogous treatments of 7- and 14-day cell cultures, respectively, from patient ZA. α, Oligonucleotide targeted to human α-globin gene as negative control (D, lane 9).
Similarly to patient DD, high levels of correct β-globin mRNA also were generated by oligonucleotide treatment of cells from the compound heterozygous patient, ZA (IVS2–745/IVS2–1), after 7 and 14 days of culture (Fig. 5 C and D, band of 231 nucleotides). However, additional products generated by RT-PCR differed in the two patients. Aberrantly spliced IVS2–745 mRNA was not detectable in ZA and, instead, a 278-nt product became apparent. This band is caused by the cryptic splice site activated by the IVS2–1 mutation, 47 nucleotides into intron 2 (27). Furthermore, as especially obvious in a 14-day culture (Fig. 5D), increased production of correct β-globin mRNA led to a concomitant decrease in the intensity of the 278-nt band (Fig. 5D, lanes 3–8). The reasons for the latter result are not entirely clear because it seems unlikely, although not impossible, that transcription and processing of the IVS2–1 allele affected or was affected by the expression of the IVS2–745 allele.
For analysis of oligonucleotide-induced hemoglobin production in cells from IVS2–745 patients, the cultures at day 8 and 12 were treated with 45 μM 745–25 morpholino oligomer and incubated for an additional 3 days. Analysis of protein lysates from these cultures showed significant levels of the Hb A in both IVS2–745 patients (Fig. 6). As in the IVS2–654 case, the expression of Hb A repressed the levels of Hb F.
Figure 6.
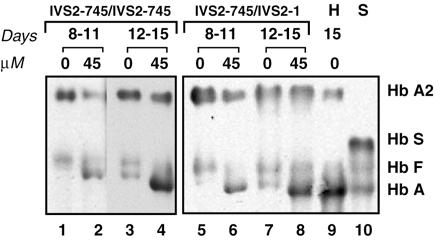
Immunoblot of hemolysate of cultured mononuclear cells from patient DD (lanes 1–4) and ZA (lanes 5–8) probed with anti-human hemoglobin antibody. The concentration of the 745–25 mer antisense oligonucleotide is indicated above the lanes. Lanes: 1–2 and 5–6, hemolysates from cells transfected on day 8 and harvested on day 11; lanes 3–4 and 7–8, hemolysates from cells transfected on day 12 and harvested on day 15; lane 9, hemolysate of mononuclear cells from an IVS2–745 heterozygote subject harvested on day 15 of culture (H); and lane 10, hemoglobin standards (S).
The extent of correction of Hb A synthesis by the oligonucleotide was estimated by comparison of the Hb A level in the treated cells to that in 15-day cultured mononuclear cells from an IVS2–745 carrier (Fig. 6, lane 9). The ratios of hemoglobin A bands to Hb A2 bands (used as internal lane loading control) in lanes 8 and 9 of Fig. 6 showed that the amount of Hb A in the oligonucleotide-treated cells from patient ZA (lane 8) was ≈30% of that in the carrier (lane 9).
Time Course of β-Globin mRNA and Hb A Repair.
The persistence of Hb A expression after a single treatment with 45 μM 745–25 morpholino oligomer was tested. The cells treated at day 8 of culture exhibited a high level of β-globin mRNA 24 h after treatment (Fig. 7, lower panel, lane 6) whereas Hb A appeared 2 days later, at day 11 (Fig. 7 Upper, lane 7). This result indicated that after the oligonucleotide treatment the cells were unharmed and that the expected course of erythroid differentiation continued. Furthermore, high levels of both Hb A and β-globin mRNA persisted even after 9 days, i.e., at day 17 of cell culture. Because the half-life of wild-type β-globin mRNA is between 30 and 50 h (28), its presence at day 17 suggested that the morpholino oligomers persisted intracellularly at a sufficient concentration to effect correction of splicing.
Figure 7.
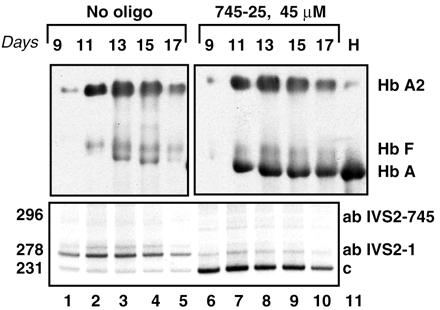
Time course of restoration of correct splicing and Hb A synthesis in mononuclear cells from patient ZA. Mononuclear cells on day 8 of culture were transfected with 45 μM 745–25 mer antisense oligonucleotide and incubated for 1–9 subsequent days with no additional oligonucleotide. Number of days after oligonucleotide treatment is indicated at the top (Upper); other designations as in Fig. 3 and 5. Lanes: 6–10, oligonucleotide-treated cells, lanes 1–5, no oligonucleotide controls. (Upper) Immunoblot of hemolysate of mononuclear cells probed with anti-human hemoglobin antibody. (Lower) Corresponding RT-PCR assay.
Detection of Hemoglobin A by Cytoimmunofluorescence in Erythroid Cells from IVS2–745 Patients.
To asses the expression of Hb A at the cellular level, cells from IVS2–745 patients were immunostained with anti-human β-globin mAb followed by anti-mouse FITC-conjugated secondary antibody. The crossreactivity of anti-human β-globin mAb with Hb E precluded this analysis in the IVS2–654/βE material. Clear cytoplasmic hemoglobin staining in a large number of nucleated cells was easily detectable in the material from both DD and ZA patients (Fig. 8, panels 6 and 8, respectively). The staining is specific for human Hb A because cells not treated with the oligonucleotide (Fig. 8, panel 2) or oligonucleotide-treated cells probed with nonimmune mouse IgG1 (Fig. 8, panel 4) were negative.
Figure 8.
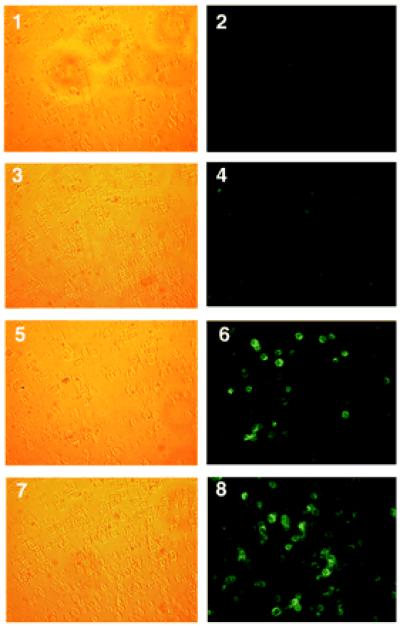
Cytoimmunofluorescent staining of Hb A in oligonucleotide-treated cells from patients DD (panels 1–6) and ZA (panels 7–8). Cells were treated with 45 μM 745–25 mer antisense oligonucleotide on day 12 of culture and incubated for additional 3 days. Panels 1, 3, 5, and 7, phase contrast; panels 2, 4, 6, and 8, fluorescence. Panels 1 and 2, no oligonucleotide controls incubated with anti-human β-globin mAb; panels 3 and 4, oligonucleotide-treated cells incubated with nonimmune mouse IgG1; and panels 5–8, oligonucleotide-treated cells incubated with anti-human β-globin mAb.
Discussion
We have shown restoration of Hb A synthesis in erythroid progenitor cells isolated from peripheral blood of thalassemic patients by their treatment with morpholino oligomers targeted to the aberrant splice sites in mutant β-globin pre-mRNAs. The restoration was accomplished for β-thalassemias with three different genotypes: IVS2–654/βE, IVS2–745/IVS2–745, and IVS2–745/IVS2–1 and in two or three separate blood samples from each patient. A single treatment with morpholino oligomers of cells cultured for 8 days restored correct β-globin mRNA, which persisted for additional 9 days (Fig. 7, lower panel, lanes 6–10). Three days post-treatment the nucleated erythroid cells became hemoglobinized, indicating translation of the newly generated β-globin mRNA and formation of Hb A and suggesting that under appropriate conditions the repaired cells may progress into mature erythrocytes. Because the lifespan of the latter is ≈120 days, the effects of the oligonucleotide treatment could be quite prolonged. If similar results were accomplished in patients with IVS2–654 and IVS2–745 thalassemias, their clinical status could improve.
The morpholino analogs of oligonucleotides have numerous desirable characteristics as modifiers of splice site selection and in model systems performed better than the oligonucleotides with other backbones (22). However, their free uptake into the erythropoietic cells was limited and even syringe loading of the cells, which entails mechanical disturbance of the cell membrane (see Materials and Methods), required high concentrations of the compounds. Note, however, that among close to 200 possible modifications of oligonucleotides (29) only a few have been tested in vivo and in clinical trials (30, 31). Furthermore, even oligonucleotides poorly taken up by the cells may be efficiently delivered by carriers such as cationic lipids or dendrimers (32). Thus, although delivery as well as in vivo bioavailability and pharmacodynamics of antisense oligonucleotides constitute serious impediments to their successful clinical application, they do not seem insurmountable. Importantly, the results reported here indicate that when the antisense compounds targeted to the aberrant splice sites are delivered to human erythroid progenitors, the targets for antisense therapy, these cells will respond as expected leading to significant restoration of hemoglobin production. Experiments on in vivo delivery of oligonucleotides in a mouse model of IVS2–654 thalassemia (23) are in progress (H.S., unpublished data).
The correction of splicing by morpholino oligomers was dose-dependent and sequence-specific for both IVS2–654 and IVS2–745 thalassemias but IVS2–745 pre-mRNA splicing was corrected more efficiently than that of IVS2–654 pre-mRNA. These results are consistent with our previous studies in model cell culture systems (21) and suggest that the sensitivities of the targeted splice sites in the IVS2–654 and IVS2–745 pre-mRNAs differ. The differences most likely reflect the nature of the interactions of the spliceosomes with the pre-mRNAs, which, in turn, must be responsible for the β0 and β+ status of IVS2–654 and IVS2–745 thalassemias (10, 14), respectively.
IVS2–654, IVS2–745 and a rare mutant, IVS2–705 (15), all activate the same 3′-cryptic splice site. Similarly, a common cryptic splice site is activated in Hb E, IVS1–5, and IVS1–6 thalassemias (14, 16, 17). An oligonucleotide targeted to a branch point in intron 1 of the β-globin gene was found effective in cell-free extracts in correction of splicing of pre-mRNA from a common thalassemic mutation, IVS2–110 (18). Thus, only three oligonucleotides may be needed in treatment of most of splicing mutations in thalassemia. Nevertheless, this approach is not applicable to β-thalassemias with other genotypes and is limited in comparison to gene therapy, which may replace or supplant any mutant with a correct gene. However, antisense approach has several advantages. Antisense oligonucleotides correct splicing of pre-mRNA that is properly transcribed from the β-globin gene in its natural chromosomal environment and controlled by the native locus control region. This precludes the possibility of over- or inappropriate expression of β-globin mRNA, an important consideration in treatment of hemoglobinopathies. Second, the approach offers a pharmacological treatment, easier to implement than gene therapy. Third, the treatment may be easily stopped if any undesirable effects are observed. Note that modification of splicing by antisense oligonucleotides is also applicable to other genetic disorders (33, 34) and to diseases, such as certain cancers that may respond to alteration of alternative splicing (ref. 35 and D. Mercatante and R.K., unpublished data).
Acknowledgments
We are grateful to the patients YC, DD, and ZA and their parents for donating the blood samples. We thank Drs. Wanla Kulwichit and Anna Astriab for their help in cytoimmunostaining and Elizabeth Smith for technical assistance. We thank Dr. Piero Pucci for the α- and β-globin samples. G.L. was supported in part by the fellowship from Consiglio Nazionale delle Ricerche, Italy. This work was supported by Grant HL-51940, National Heart, Lung and Blood Institute, National Institutes of Health (to R.K.).
Abbreviations
- Hb
hemoglobin
- pre-mRNA
precursor mRNA
- RT-PCR
reverse transcription-PCR
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Cohen A, editor. Cooley's Anemia: Progress in Biology and Medicine. Bethesda, MD: National Institutes of Health, National Heart, Blood and Lung Institute; 1995. [Google Scholar]
- 2.Schwartz E, Benz E J, Jr, Forget B G. In: Hemalotogy, Basic Principles and Practice. Hoffman R, Benz E J, Shattil S J, Furie B, Cohen H J, editors. New York: Churchill Livingstone; 1995. pp. 586–610. [Google Scholar]
- 3.Olivieri N F, Weatherall D J. Hum Mol Genet. 1998;7:1655–1658. doi: 10.1093/hmg/7.10.1655. [DOI] [PubMed] [Google Scholar]
- 4.Olivieri N F. N Engl J Med. 1999;341:99–109. doi: 10.1056/NEJM199907083410207. [DOI] [PubMed] [Google Scholar]
- 5.Fucharoen S, Winichagoon P. Curr Opin Hematol. 2000;7:106–112. doi: 10.1097/00062752-200003000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Perrine S P, Dover G H, Daftari P, Walsh C T, Jin Y, Mays A, Faller D V. Br J Haematol. 1994;88:555–561. doi: 10.1111/j.1365-2141.1994.tb05073.x. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Emery D W, Fernandez M, Han H, Stamatoyannopoulos G. Blood. 1999;93:2208–2216. [PubMed] [Google Scholar]
- 8.Russell J E, Liebhaber S A. Blood. 1998;92:3057–3063. [PubMed] [Google Scholar]
- 9.Huisman T H. Br J Haematol. 1990;75:454–457. doi: 10.1111/j.1365-2141.1990.tb07781.x. [DOI] [PubMed] [Google Scholar]
- 10.Cheng T C, Orkin S H, Antonarakis S E, Potter M J, Sexton J P, Li A, Kazazian H H., Jr Proc Natl Acad Sci USA. 1984;81:2821–2825. doi: 10.1073/pnas.81.9.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang S Z, Zeng F Y, Ren Z R, Lu Z H, Rodgers G P, Schechter A N, Zeng Y T. Br J Haematol. 1994;88:541–546. doi: 10.1111/j.1365-2141.1994.tb05071.x. [DOI] [PubMed] [Google Scholar]
- 12.Busslinger M, Moschonas N, Flavell R A. Cell. 1981;27:289–298. doi: 10.1016/0092-8674(81)90412-8. [DOI] [PubMed] [Google Scholar]
- 13.Fukumaki Y, Ghosh P K, Benz E J, Jr, Reddy V B, Lebowitz P, Forget B G, Weissman S M. Cell. 1982;28:585–593. doi: 10.1016/0092-8674(82)90213-6. [DOI] [PubMed] [Google Scholar]
- 14.Treisman R, Orkin S H, Maniatis T. Nature (London) 1983;302:591–596. doi: 10.1038/302591a0. [DOI] [PubMed] [Google Scholar]
- 15.Dobkin C, Bank A. J Biol Chem. 1985;260:16332–16337. [PubMed] [Google Scholar]
- 16.Weatherall D J, Higgs D R. The Haemoglobinopathies, Bailliere's Clinical Haematology. Vol. 6. London: Saunders; 1993. [Google Scholar]
- 17.Miller D R, Baehner R L, editors. Blood Diseases of Infancy and Childhood. St. Louis: Mosby; 1995. [Google Scholar]
- 18.Dominski Z, Kole R. Proc Natl Acad Sci USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sierakowska H, Sambade M J, Agrawal S, Kole R. Proc Natl Acad Sci USA. 1996;93:12840–12844. doi: 10.1073/pnas.93.23.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sierakowska H, Montague M, Agrawal S, Kole R. Nucleosides & Nucleotides. 1997;16:1173–1182. [Google Scholar]
- 21.Sierakowska H, Sambade M J, Schumperli D, Kole R. RNA. 1999;5:369–377. doi: 10.1017/s135583829998130x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmajuk G, Sierakowska H, Kole R. J Biol Chem. 1999;274:21783–21789. doi: 10.1074/jbc.274.31.21783. [DOI] [PubMed] [Google Scholar]
- 23.Lewis J, Yang B, Kim R, Sierakowska H, Kole R, Smithies O, Maeda N. Blood. 1998;91:2152–2156. [PubMed] [Google Scholar]
- 24.Summerton J, Weller D. Antisense Nucleic Acid Drug Dev. 1997;7:187–195. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 25.Partridge M, Vincent A, Matthews P, Puma J, Stein D, Summerton J. Antisense Nucleic Acid Drug Dev. 1996;6:169–175. doi: 10.1089/oli.1.1996.6.169. [DOI] [PubMed] [Google Scholar]
- 26.Chen I T, Chasin L A. Mol Cell Biol. 1993;13:289–300. doi: 10.1128/mcb.13.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treisman R, Proudfoot N J, Shander M, Maniatis T. Cell. 1982;29:903–911. doi: 10.1016/0092-8674(82)90452-4. [DOI] [PubMed] [Google Scholar]
- 28.Steinberg M H, Benz J., Jr . In: Hemalotogy, Basic Principles and Practice. Hoffman R, Benz E J, Shattil S J, Furie B, Cohen H J, editors. New York: Churchill Livingstone; 1995. pp. 458–468. [Google Scholar]
- 29.Freier S M, Altmann K H. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agrawal S, Kandimalla E R. Mol Med Today. 2000;6:72–81. doi: 10.1016/s1357-4310(99)01638-x. [DOI] [PubMed] [Google Scholar]
- 31.Marcusson E G, Yacyshyn B R, Shanahan W R, Jr, Dean N M. Mol Biotechnol. 1999;12:1–11. doi: 10.1385/MB:12:1:1. [DOI] [PubMed] [Google Scholar]
- 32.Yoo H, Sazani P, Juliano R L. Pharm Res. 1999;16:1799–1804. doi: 10.1023/a:1018926605871. [DOI] [PubMed] [Google Scholar]
- 33.Friedman K J, Kole J, Cohn J A, Knowles M R, Silverman L M, Kole R. J Biol Chem. 1999;274:36193–36199. doi: 10.1074/jbc.274.51.36193. [DOI] [PubMed] [Google Scholar]
- 34.Wilton S D, Lloyd F, Carville K, Fletcher S, Honeyman K, Agrawal S, Kole R. Neuromuscul Disord. 1999;9:330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 35.Taylor J K, Zhang Q Q, Wyatt J R, Dean N M. Nat Biotechnol. 1999;17:1097–1100. doi: 10.1038/15079. [DOI] [PubMed] [Google Scholar]