

Abstract
A significant proportion of familial breast cancers cannot be explained by mutations in the BRCA1 or BRCA2 genes. We applied a strategy to identify predisposition loci for breast cancer by using mathematical models to identify early somatic genetic deletions in tumor tissues followed by targeted linkage analysis. Comparative genomic hybridization was used to study 61 breast tumors from 37 breast cancer families with no identified BRCA1 or BRCA2 mutations. Branching and phylogenetic tree models predicted that loss of 13q was one of the earliest genetic events in hereditary cancers. In a Swedish family with five breast cancer cases, all analyzed tumors showed distinct 13q deletions, with the minimal region of loss at 13q21-q22. Genotyping revealed segregation of a shared 13q21 germ-line haplotype in the family. Targeted linkage analysis was carried out in a set of 77 Finnish, Icelandic, and Swedish breast cancer families with no detected BRCA1 and BRCA2 mutations. A maximum parametric two-point logarithm of odds score of 2.76 was obtained for a marker at 13q21 (D13S1308, θ = 0.10). The multipoint logarithm of odds score under heterogeneity was 3.46. The results were further evaluated by simulation to assess the probability of obtaining significant evidence in favor of linkage by chance as well as to take into account the possible influence of the BRCA2 locus, located at a recombination fraction of 0.25 from the new locus. The simulation substantiated the evidence of linkage at D13S1308 (P < 0.0017). The results warrant studies of this putative breast cancer predisposition locus in other populations.
Breast Cancer Linkage Consortium data on 237 breast-ovarian cancer families showed that 52% were linked to BRCA1 and 32% to BRCA2 (1). Recent reports indicate that the proportion of breast cancer families attributable to the BRCA1 and BRCA2 genes may be smaller than initially thought, especially in studies that have been based on population-based family materials. For instance, in Finnish breast cancer families with three or more affected cases, mutations of the BRCA1 gene were seen in only 10% and those of BRCA2 in only 11% of the families (2). In southern Sweden, the corresponding percentages were 23% and 11% (3). These studies suggest that in the Nordic populations, a significant proportion of familial breast cancer is not explained by the two known major susceptibility genes. Therefore, identification of additional breast cancer susceptibility genes is an important goal.
According to the two-hit model of cancer development (4), hereditary cancers arise as a result of a germ-line mutation in a recessive tumor suppressor gene, followed by the somatic deletion of the wild-type allele of the gene. Somatic deletions detected from tumor tissues of patients with a genetic predisposition therefore may pinpoint those loci that harbor recessive germ-line mutations. Such somatic deletions detected by comparative genomic hybridization (CGH) recently were used to assign the locus for the Peutz-Jeghers' cancer syndrome, an intestinal hamartoma-carcinoma syndrome (5, 6). In our recent study of tumor specimens from families with BRCA1 and BRCA2 carriers (7), deletion of the 13q region (the site of the BRCA2 gene) was the most common alteration (73%) in tumors from BRCA2 cancers. This finding suggests that the wild-type BRCA2 allele in tumor cells often may be inactivated by a large somatic deletion. The challenge of this molecular pathological approach lies in how to distinguish primary deletion events involved in cancer initiation from the numerous deletion events that arise during cancer progression.
Here, we used CGH to identify chromosomal regions that may harbor novel breast cancer predisposition genes. We performed CGH analyses of 61 tumor tissues from 37 non-BRCA1/BRCA2 (BRCAX) breast cancer families. Distinction of early genetic events was facilitated by the application of two complementary mathematical tree models to analyze the CGH data (8, 9). In addition, we searched for deletions that were shared in tumor tissues from multiple affected cases of the same family. A candidate region at 13q21-q22 that emerged from this molecular pathology approach was further evaluated by using linkage analysis.
Patients and Methods
Tumor Specimens and Families.
A total of 61 tumor specimens from 37 breast cancer families were selected for CGH analysis from the Helsinki University Central Hospital (Finland, 24 tumors), Lund University Hospital (Sweden, 23 tumors), and University Hospital of Iceland (14 tumors). Exclusion of BRCA1 and BRCA2 deleterious germ-line mutations was performed for each kindred by single-stranded conformation polymorphism analysis alone or combined with heteroduplex analysis, protein truncation test, or direct sequencing as well as detection of the country-specific founder mutations. In the text, this group will be called BRCAX cancers. All tumors originated from families with three or more breast cancer patients: 10 families had three affecteds, 16 had four affecteds, seven had five affecteds, and four had six or more affecteds. Seven of the families included a woman diagnosed with ovarian cancer. Average age at diagnosis of the patients in the CGH analysis set was 52.5 years (range 33 to 80), the average age of all affecteds in the families was 54.2 years (range 29 to 87).
For linkage analysis, samples from 77 multiplex (57 Finnish, 12 Icelandic, and eight Swedish) families were genotyped. Twenty-three of these were families included in the CGH analysis and from which blood samples from multiple affecteds were available. The families were identified through the oncology clinics of the Helsinki, Tampere, and Lund university hospitals and the Department of Pathology of the University Hospital of Iceland. Blood samples were collected from living family members who gave an informed consent. Paraffin tissue blocks were obtained from deceased cancer patients when available. DNA was extracted from blood with the PureGene (Gentra Systems) kit according to manufacturer's protocol. Paraffin samples were processed as described below. Exclusion of BRCA1 and BRCA2 germ-line mutations was performed as described above. The study was approved by the Ministry of Social Affairs and Health in Finland, local ethics committees of the Helsinki, Tampere, Lund, and Iceland university hospitals, as well as by the National Institutes of Health.
CGH.
CGH was performed essentially as described (7, 10) from 61 hereditary, non-BRCA1, and BRCA2-associated cancers. DNAs from Sweden (23 cases) came from freshly frozen tumors, whereas the remaining 38 were from formalin-fixed paraffin-embedded archival samples. DNA was extracted by using a QIAmp tissue kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. In seven paraffin-embedded cases, preamplification of the DNA using degenerate oligonucleotide-primed PCR methodology was necessary, as described in a previous study (11). The CGH results in this study were compared with those of our previous studies of 55 sporadic, primary breast cancers (10) as well as 21 cancers from BRCA1 and 15 BRCA2 mutation carriers (7).
Selection of Nonrandom Events.
Many of the gains and losses observed by CGH are probably random and not related to the progression of the tumor. In data sets from unselected and hereditary cancers, random events may recur multiple times. The events here all are based on copy number aberrations, although the models do not depend on what type of data it is. The event is binary, i.e., either “present” or “absent.” The methods have been used in the past to model, not only CGH data, but also breakpoint data, translocations, and gene expression data. To select which recurrent events are most likely to be nonrandom (i.e., truly related to the cause of tumor development and progression), we used a well-established method of Brodeur et al. (12). This method requires that the prior probabilities of events are proportional to the size of the chromosomal region involved; for this purpose we used chromosome arm size estimates from Morton (13).
Construction of Tree Models of Events.
To find possible hypotheses as to which events are early/late and which pairs of events may have cause-and-effect relationships, we used two different methods recently developed by Desper et al. (8, 9). The essential goal of the method of Desper et al. (8) is to find a “tree” structure among the set of events selected as nonrandom, adding one event called the root, which represents normal diploid cells in the target organ for the cancer development. A tree structure connects the events by “edges” in such a way that there is a unique path of edges from the root to each other event. The intuition behind such a tree structure is that: (i) events closer to the root are earlier events, (ii) events connected by an edge may have a cause-and-effect relationship, and (iii) events clustered in a subtree may define a genetic subclass of tumors.
Desper et al. (8) defined a probability space of tree models, in which each tree model T can be used to generate a distribution D(T) from which one can sample tumors. By this, we mean that each sample from D(T) is a set of gains and losses. The problem of selecting a tree is to take the observed real samples and find a tree structure whose associated distribution may have generated these samples.
Desper et al. (8) suggested how the observed probabilities of events and observed joint probabilities of pairs of events could be used to assign a weight to each possible edge between pairs of events. For the edge directed from i to j, the weight function they suggested is:
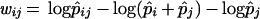 |
where p̂i is the observed probability of event i and logp̂ij is the observed joint probability of i and j. The weight function is not symmetric in i and j because the edges selected should be directed away from the root, and so the edges from i to j and from j to i should not get the same weight in most cases. If there are n events including the root, this weight function defines weights for n*(n-1) possible edges, but a tree has only n-1 edges. Desper et al. (8) used a method from computer science called (selection of) a “maximum-weight branching” (14–17) that can efficiently select a tree of directed edges, such that all edges are consistently directed away from the root, and the sum of the n-1 edge weights is maximized. To connect the maximum-weight branching back to the probabilistic model of tumor formation, Desper et al. (9) proved that if the observed probabilities of events and pairs of events accurately estimate the true probabilities, then the maximum branching tree has the same edge set as the true underlying tree model.
In Desper at al (9). a different version of the problem was considered where the desired tree is a phylogenetic tree in which the visible CGH events are all leaves of the tree. The internal nodes are considered to be invisible events. For this method and the data set herein, we proceeded as follows.
(i) A distance-matrix was generated by using the transformation:
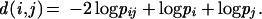 |
(ii) The distance matrix was fed as input to three different programs used to fit trees to distance matrices. We used the Fitch and Neighbor programs from the phylip software package (ref. 18, see also the phylip homepage http://evolution.genetics.washington.edu/phylip.html).
(iii) For each topology obtained in step ii, the edge-weights were optimized by using a linear program. This resulted in a distance matrix for each topology. The fitness of each topology was measured by comparing the output tree metric with the input metric, under L1, L2, and L∞ norms.
(iv) In the analysis of the trees for the hereditary cancers, the optimal tree was the weighted Fitch tree. It was superior to the other output trees when measured by all three of the norms used in step iii.
Linkage Analysis.
Seventy-seven families were genotyped by using 23 genetic markers (Genome Database, http://gdbwww.gdb.org). Only females diagnosed with breast cancer were coded as affected. Women with ovarian cancer were coded as unknown, whereas all other females were coded unaffected. All males were classified as unknown. In the 57 Finnish families, there were 222 affecteds (mean 3.9 per family, range 2 to 7), and 68% of the affecteds were genotyped. In the 12 Icelandic families, there were 75 affecteds (mean 6.3 per family, range 4 to 11), and 67% of affecteds were genotyped. In eight Swedish families, there were 37 affecteds (mean 4.6 per family, ranging from 3 to 7), and 86% of affecteds were genotyped. Genotyping was performed in three different facilities (National Human Genome Research Institute, Lund, and Reykjavik) essentially as described by Smith et al. (19).
Genotyping data were first checked for inconsistency by using pedcheck (20). Model-dependent logarithm of odds (lod) score linkage analysis was performed by using fastlink software (21–24). The genetic model was autosomal dominant inheritance, of a disease allele with a population frequency of 0.0033 (25). We used age-dependent penetrances for mutation carriers and phenocopies derived by Easton et al. (26) from the Cancer and Steroid Hormone Study (26), which was a reasonable compromise for all three countries. Locus heterogeneity was tested by using homog2 (for BRCA2 and putative BRCAX) and homog3r (for BRCA1 and putative BRCAX) (27). Model-independent affected sibpair analysis was performed by using the sibpal program in the Statistical Analysis for Genetic Epidemiology 3.1 (S.A.G.E.) package. Identity by descent (IBD) from each population was pooled, and overall P values were derived based on pooled IBD. We estimated country-specific allele frequencies from independent individuals, married-in spouses, and unrelated controls (30 from Finland, 50 from Iceland, 50 from Sweden) by sib-pair (http://www.qimr.edu.au/davidD/davidd.html). Multipoint linkage analyses were performed by using fastlink (22–24) and genehunter (28). Published map distances from Généthon (http://www.genethon.fr/genethon_en.html) between the marker loci were used in these analyses. Multipoint heterogeneity lod scores (Hlod) were calculated by using homog (27).
The statistical significance of the peak two-point lod score with marker D13S1308 was assessed by simulation, using the principles outlined in ref. 27, but adding two variations needed for this data set. Usually, one simulates n unlinked replicates, computes lod scores in each replicate, and counts how many replicates have a score exceeding the true score. For the analysis herein, we need to note the following. None of 3,000 replicates above the true score yields a P value of < 0.0010; one of 3,000 replicates above the true score yields a P value of < 0.0017. When the X-chromosome is targeted for linkage analysis, P < 0.01 is sufficient evidence to declare linkage because of the prior evidence targeting X and the reduced multiple testing, as compared with genomewide analysis (27). Because chromosome 13 is smaller than chromosome X (13), the appropriate threshold for significance is no lower than P < 0.01.
We used the simulation software package fastlink (21, 23, 29) to generate replicates. To evaluate lod scores in each replicate we used the linkage analysis package fastlink, as for the real data. We used the same penetrance classes and country-specific allele frequencies in the simulations as with the real data. To make a combined ith-replicate for three countries, we pooled together the ith replicate from each.
Because BRCA2 also is located on chromosome 13, the simulations must allow for some of the 13q21-linked families to have breast and ovarian cancer cases caused by undetected BRCA2 mutations and not by a novel locus. We estimated that the recombination fraction between D13S1308 and BRCA2 is 0.25. We generated a pool of “linked” replicates in which the simulated marker is linked at a recombination fraction of 0.25 from the disease gene. When generating linked replicates, all individuals with ovarian cancer were coded affected.
To generate a random three-country replicate in which F Finnish families, I Icelandic families, and S Swedish families are linked to BRCA2, we proceeded as follows. We chose F distinct random numbers in {1, … , 57}, I distinct random numbers in {1, … , 12}, S distinct random numbers in {1, … , 8}. For each random number chosen we replaced the unlinked copy of that family with a linked copy from the pool of linked replicates. The selection of linked families was without replacement. To evaluate the lod score of a three-country replicate, we first summed the lod scores over the three countries at each θ up to 0.1, then maximized over θ.
Results
The Most Common Genetic Aberrations in BRCAX Tumors by CGH.
Losses in BRCAX tumors were most often seen at chromosome arms 13q (56%), 6q (41%), 11q (30%), 9p (26%), and Xq (26%). The 10 most common somatic genomic alterations in these cancers, and in other types of familial and sporadic breast cancers analyzed previously in our laboratory, are shown in Table 1. Gains in BRCAX tumors were clustered at regions that are also typical to unselected breast cancers, such as 1q (46%), 17q (39%), 8q (36%), and 16p (34%), whereas losses were more evenly distributed around the genome. In BRCAX cancers, the total number of copy number alterations (CNA) was 9.4 ± 5.5 (range 1 to 19), not significantly differing from the sporadic set (7.7 ± 5.1). However, the BRCAX-associated tumors did have a lower number of CNAs than BRCA1-associated (12.2 ± 5.3, n = 21) and BRCA2-associated (12.5 ± 4.9, n = 15) tumors (P = 0.02 according to one-way ANOVA).
Table 1.
Ten most common somatic alterations in BRCAX cancers (percentage of cases), as compared to other hereditary and unselected breast cancers by CGH
| Location | BRCAX | BRCA1 (7) | BRCA2 (7) | Unselected (10) |
|---|---|---|---|---|
| Loss 13q21–q31 | 56 | 55 | 73* | 26 |
| Gain 1q22–q32 | 46 | 55 | 53 | 66 |
| Loss 6q16–q24 | 41 | 27 | 60 | 18 |
| Gain 17q23 | 39 | 46 | 87 | 18 |
| Gain 8q23–q24 | 36 | 73 | 53 | 49 |
| Gain 16p13–p12 | 34 | 27 | 40 | 38 |
| Loss 11q14–q24 | 30 | 23 | 53 | 18 |
| Gain 19q | 30 | 18 | 40 | 20 |
| Loss 9p23–p21 | 26 | 27 | 40 | 20 |
| Loss Xq | 26 | 40 | 47 | 20 |
Target area 13cen–q21.
Construction of Tree Models for Breast Cancer Progression.
The method of Brodeur et al. (12) selected the following events as nonrandom in BRCAX cases: +1q, +8q, +16p, +17q, +19p, +19q, +20q, −6q, −8p, −9p, −11q, and −13q. Brodeur's method used in the tree models takes into account the relative sizes of the 41 chromosome arms, whereas the frequency counts in the previous paragraph ignore this information. In sporadic cancers, the selected events were: +1q, +5p, +8q, +16p, +17q, +19p, +19q, +20q, −8p, −16q, and −17p. These two sets of tumors have eight events in common: +1q, +8q, +16p, +17q, +19p, +19q, +20q, and −8p. Interestingly, several gains are common to both data sets, but only a few losses were selected as nonrandom events in both.
Next, tree models were constructed (Fig. 1 A and B) for the BRCAX cancers according to Desper et al. (8, 9). The loss of 13q was selected as being an early event in the progression of BRCAX tumors with both branching and phylogenetic (distance matrix) trees. In contrast, 6q loss was located far from the root of both tree types, suggesting that it may not be an early event in the progression of these tumors.
Figure 1.
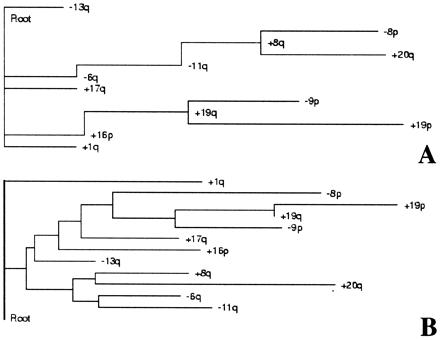
A schematic representation of results from the tree analysis of nonrandom genomic alterations of BRCAX breast carcinomas. The root represents a normal diploid mammary cell, whereas only events above the 95th percentile of the distribution are shown (see text for details). (A) Shown is how the observed probabilities of events and observed joint probabilities of pairs could be used to assign a weight to each possible “edge” between pairs and events. The edge represents a putative cause-and-effect relationship between events, whereas clusters of events may define a genetic subclass of tumors. (B) The result obtained from the best fitting distance matrix, weighted Fitch tree (see text for details). Both independent probabilistic approaches thus led to the following finding: The loss of chromosome 13 seems to be an early event in the pathogenesis of BRCAX breast cancers, and it is not strongly associated with other early or late events.
13q Losses in Tumor Tissues and Germ-Line Haplotype Sharing in a BRCAX Breast Cancer Family.
Compelling evidence implicating 13q loss as an early event in the BRCAX tumors came from one large family (L5), where all five samples analyzed from different patients showed a deletion at 13q, with the consensus region of deletion at 13q21-q22 (Fig. 2). Loss of 13q was the only genetic alteration common to all five tumors from this family. Several other families showed individual tumors with the same narrow region of loss. Loss at 13q was common in samples from each country (Finland 13/24, Sweden 13/23, and Iceland 8/14).
Figure 2.
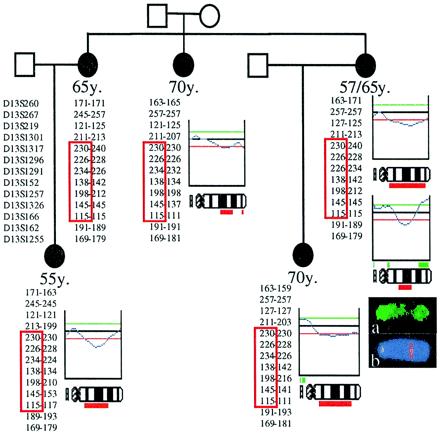
Pedigree Lund 5 shown with the CGH profiles of chromosome 13, demonstrating loss of 13q21-q22 in all five tumors from the four affected individuals. The haplotypes with markers from 13q are shown next to the CGH profiles (the marker map is located on the left). Markers located near BRCA2 were not shared between the affected individuals (D13S260 and D13S267). At 13q21-q22, however, all five affected women shared a 7 cM haplotype (red box). Below the CGH profile of a bilateral breast cancer is the original CGH image (FITC hybridization with tumor DNA). The interstitial 13q21-q22 loss can be observed as an absence of FITC signal (green color) in the middle of the chromosome (a). Hybridization with a Texas red-labeled yeast artificial chromosome probe for marker D13S1257 demonstrates that the region of the shared haplotype coincided with the region of deletion by CGH (b).
To test whether losses of the 13q21-q22 region were selected for because this region harbors a germ-line alteration predisposing to breast cancer, we genotyped genetic markers along the 13q region in the germ-line DNA of the L5 family members. A 7.3 cM haplotype was shared between all five affected cases in this family with markers along 13q. The region of sharing was apparent between markers D13S1317 and D13S166. Markers at the BRCA2 region were not shared (Fig. 2). A metaphase fluorescence in situ hybridization experiment localized a yeast artificial chromosome probe for a shared genetic marker (D13S1257) to 13q22. This haplotype sharing region was therefore identical with the region of loss in the tumors by CGH (Fig. 2).
Linkage Analysis.
Linkage analysis was carried out in 77 breast cancer families from Finland, Sweden, and Iceland. We found that three markers, D13S1308, D13S1257 and D13S791, had two-point lod scores > 1 in the Finnish population with a peak two-point lod score of 2.89 at D13S1308 with θ = 0.04. After combination with the Swedish and the Icelandic data, six markers within a 10 cM region (D13S1313 to D13S162) had two-point lod scores greater than 1 (Table 2). The peak lod score was 2.76, θ = 0.10 with marker D13S1308. There was no evidence for sex dependence of the recombination fraction.
Table 2.
Combined lod scores from the 77 families at several values of theta for markers on 13q
| Marker | cM | lod score at θ
|
|||
|---|---|---|---|---|---|
| 0.00 | 0.05 | 0.10 | 0.20 | ||
| D13S260 | −8.24 | −4.81 | −2.92 | −1.01 | |
| D13S1701 | 1.6 | −4.36 | −2.48 | −1.43 | −0.39 |
| D13S267 | 0 | −2.35 | −1.16 | −0.56 | −0.04 |
| D13S1301 | 20.3 | −1.75 | −0.69 | −0.16 | 0.23 |
| D13S1313 | 0.8 | 0.94 | 1.13 | 1.18 | 0.98 |
| D13S1317 | 1.9 | −1.87 | −0.18 | 0.58 | 0.96 |
| D13S1262 | 0 | −4.01 | −2.03 | −1.09 | −0.21 |
| D13S275 | 0.8 | −0.24 | 0.7 | 0.98 | 0.87 |
| D13S1296 | 0.8 | −0.27 | 0.79 | 1.13 | 1.05 |
| D13S1308 | 0 | 1.61 | 2.56 | 2.76 | 2.19 |
| D13S1291 | 0.5 | −2.64 | −0.85 | −0.03 | 0.52 |
| D13S152 | 2.6 | −5.58 | −2.12 | −0.67 | 0.41 |
| D13S1257 | 0 | −0.4 | 0.64 | 1.14 | 1.26 |
| D13S745 | 0 | 0.63 | 0.86 | 0.89 | 0.68 |
| D13S791 | 0 | −1.55 | 0.36 | 0.99 | 1.09 |
| D13S1326 | 0 | −3.34 | −1.2 | −0.28 | 0.32 |
| D13S1249 | 0.7 | −0.45 | 0.16 | 0.47 | 0.59 |
| D13S166 | 0 | −4.93 | −2.03 | −0.7 | 0.39 |
| D13S269 | 1.0 | −1.66 | −0.2 | 0.5 | 0.85 |
| D13S162 | 1.4 | −1.13 | 0.68 | 1.39 | 1.55 |
| D13S1306 | 1.2 | −3.01 | −0.98 | −0.07 | 0.51 |
| D13S160 | 1.8 | −2.26 | −0.5 | 0.18 | 0.47 |
| D13S1255 | 1.6 | −2.87 | −1.08 | −0.16 | 0.48 |
The markers D13S260, D13S1701 and D13S267 flank the BRCA2 gene. The peak lod score of 2.76 was seen at marker D13S1308 at θ = 0.10.
Lod scores greater than 1.0 are shown in bold.
Testing for the presence of both BRCA1-linked families and families linked to the putative BRCAX region on chromosome 13q by homog3r analyses showed no evidence of heterogeneity, indicating a low fraction of BRCA1-linked families in this data set. The homog2 analyses, testing for the presence of both BRCA2- and BRCAX-linked families in the data also did not result in significant evidence of heterogeneity (P > 0.3). This finding suggests that although there may be a few BRCA2-linked families that were not discovered in the extensive mutation detection, the proportion of such families is likely to be small.
Multipoint lod scores, using the sliding three-point (one disease locus vs. two markers) technique in fastlink, were positive (2.60) only between markers D13S1296 and D13S1308. The highest Hlod score was 3.46 with estimated proportion of linked families (α) of 0.65 in the region between D13S1296 and D13S1308, and the Hlods were positive from D13S1262 to D13S160 (Table 3). Multipoint Hlods from genehunter were positive but always considerably lower than the corresponding ones obtained from fastlink. These discrepancies are caused by the fact that many of our families were too large to be analyzed by any implementation of the Lander-Green algorithm in genehunter.
Table 3.
Multipoint (three-point) lod scores under homogeneity and heterogeneity.
| Interval within markers | Maximum multipoint lod within the interval | Maximum multipoint Hlod within the interval (α) |
|---|---|---|
| D13S260–D13S1701 | −10.72 | 0 (0) |
| D13S1701–D13S1301 | −1.27 | 0.05 (0.17) |
| D13S1301–D13S1262 | −4.16 | 0 (0) |
| D13S1262–D13S1296 | −1.26 | 1.50 (0.37) |
| D13S1296–D13S1308 | 2.60 | 3.46 (0.65) |
| D13S1308–D13S1291 | −0.52 | 1.49 (0.42) |
| D13S1291–D13S152 | −6.08 | 0.06 (0.09) |
| D13S152–D13S1257 | −4.08 | 0.76 (0.29) |
| D13S1257–D13S745 | −2.25 | 0.90 (0.39) |
| D13S745–D13S791 | −0.88 | 0.95 (0.43) |
| D13S791–D13S1326 | −5.45 | 0.16 (0.16) |
| D13S1326–D13S166 | −5.65 | 0.26 (0.16) |
| D13S166–D13S269 | −5.64 | 0.39 (0.17) |
| D13S269–D13S162 | −2.60 | 0.66 (0.25) |
| D13S162–D13S160 | −2.92 | 0.68 (0.31) |
Lod scores greater than 1.0 are shown in bold. α denotes the percentage of families linked.
Nonparametric affected sib-pair analysis also supported evidence of linkage at 13q21-q22. The number of informative affected sib-pairs ranged from 33 to 65 in the Finnish (mean 50), from two to 12 in the Icelandic (mean 7), and from 10 to 23 in the Swedish (mean 20) families. The most significant results were obtained in the Finnish families at marker D13S1257 (P = 0.003) with D13S1308 showing borderline significance (P = 0.05). In all 77 families, three markers were slightly positive: D13S1257 (P = 0.015), D13S791 (P = 0.05), and D13S269 (P = 0.04).
Tumor tissue for loss of heterozygosity analysis was available from 16 individuals from six families with evidence of linkage to 13q22. Allelic imbalance with multiple markers at 13q22 was observed in 11/16 cases (69%), whereas the frequency observed in sporadic tumors in this region is ≈25% (30). In eight of the 11 cases it was the putative wild-type allele that demonstrated loss.
A simulation experiment was carried out to validate the significance of the two-point linkage score empirically. The simulation also addressed the concern that undetected BRCA2 mutation-positive families had influenced the observed lod scores at 13q21-q22. We conservatively estimated that the upper limit of BRCA2-linked families would be 17/57 Finnish families, 1/12 Icelandic families, and 1/8 Swedish families. In all possible configurations allowing this number of families to have undetected BRCA2 mutations, the simulated lod score at D13S1308 exceeded the threshold of 2.76 (corresponding to the peak true lod score) in at most one of the 3,000 replicates. This finding corresponds to a significance level of P < 0.0017 (27). For a linkage analysis that targets a chromosome no larger than X because of substantial prior evidence for linkage, P < 0.01 is considered sufficient evidence for linkage (27).
Discussion
Identification of a candidate susceptibility locus at 13q21 for breast cancer was based on a strategy that assumed that somatic genetic changes in cancer tissues may give insights to the nature of the germ-line predisposing loci (5). For this purpose, it is critical to distinguish early, initiating genetic changes from those that are associated with cancer progression. In our analyses of BRCA1 and BRCA2 mutation-negative breast cancer families, four intersecting lines of evidence suggest that the 13q losses observed in these tumors may represent early, initiating genetic alterations. First, the overall frequency of 13q loss was about two times higher in BRCAX tumors than in control breast cancers. Second, exploring the CGH data with two different tree models placed the loss of 13q at or near the root of the hypothetical breast cancer progression pathways. Third, tumor tissues from patients belonging to the same cancer family showed the same specific deletion at 13q21-q22 and shared a germ-line haplotype at this locus. Finally, and most importantly, unselected BRCAX breast cancer families showed significant evidence of linkage to the 13q21-q22 region. Taken together, these findings support the hypothesis that the loss of 13q21-q22 is an early genetic event in the BRCAX tumors and possibly uncovers a recessive tumor suppressor gene at this locus. The findings therefore provide preliminary evidence for the location of a novel predisposition locus for breast cancer at 13q21-q22.
In the 77 families, a maximum two-point lod score of 2.76 and a peak multipoint Hlod of 3.46 (α = 0.65) were observed. These lod scores were obtained from a linkage analysis targeting a single candidate site in the genome, based on prior suggestions from CGH. This situation is in sharp contrast to genome-wide linkage scans, where hundreds of comparisons are performed, and therefore much more stringent thresholds for statistical significance need to be applied. The two-point linkage result was further evaluated by means of a simulation analysis. Based on haplotype sharing at the BRCA2 locus, we modeled that up to 17 Finnish families, one Icelandic family, and one Swedish family to be putative BRCA2 families, in which BRCA2 mutations may have been missed. This was done to evaluate the influence of putative “contaminating” BRCA2 families on the lod scores observed in the 13q21-q22 region. The simulation results suggested that the observed linkage to D13S1308 was significant at a level of P < 0.0017. It will be critically important to independently validate this genetic locus for breast cancer, especially in populations outside of the Nordic countries.
In the analysis of the somatic progression of cancer by cytogenetics, CGH, and other methods, usually only the frequency of the genetic alterations is reported. Tree models will provide substantial additional information on the associations between genetic alterations in a series of tumors, as well as on their putative sequence of appearance. Although the progression pathway involving 13q losses may be a predominant “route” by which BRCAX cancers arise, not all BRCAX tumors evolve through this pathway. This chromosomal region often is also involved in sporadic breast cancers and other types of cancers, including prostate cancer (31, 32), upper gastrointestinal cancer (33), and nasopharyngeal cancer (34), which suggests the involvement of several genes at this region in tumor progression. Most, but not all, of the families displayed loss of the wild-type allele with markers along 13q21. The possibility of multiple tumor suppressor genes at 13q may well influence the observed loss of heterozygosity pattern: a small initial deletion in the wild-type chromosome may be masked by larger deletions occurring during tumor progression. Furthermore, because of the small family size, defining individual “linked” families could not be done with absolute certainty. This may lead to an appearance of a loss of the “mutant” allele, especially as some families also may contain sporadic breast cancers. There are numerous putative candidate target genes in the 13q21-q22 region, including protocadherin 9, the serine/threonine protein kinase EMK, the human homologue of the Drosophila dachshund gene, and the ubiquitin carboxyl-terminal esterase L3 (genemap 99; http://www.ncbi.nlm.nih.gov/genemap99).
CGH was previously used to pinpoint a predisposition locus for Peutz-Jeghers syndrome (5, 6), a single-gene disorder with a defined early premalignant lesion, which was used as a starting point for the analysis. Mutations in the gene LKB1/STK11—located at the locus assigned by the targeted linkage analysis—subsequently were identified in Peutz-Jeughers' families (35, 36). Based on the present study, we believe that the CGH approach also will be powerful for the analysis of several types of cancers with a complex genetic basis. CGH also gave suggestions on the biological properties of the BRCAX tumors. The total number of genetic alterations per tumor in the BRCAX cases was nearly identical with that seen in unselected sporadic cases and about half of that seen in BRCA1 and BRCA2 tumors. The degree of genetic instability in the BRCAX tumors therefore may be less prominent than in BRCA1- and BRCA2-associated tumors, where the loss of function of the DNA repair pathways has been suggested to play a pathogenetic role (37).
In conclusion, our results suggest that somatic genetic deletions taking place in tumor tissues from cancer families may provide a powerful approach to define candidate cancer predisposition loci, assuming that one can distinguish primary genetic alterations from secondary ones. A mathematical tree analysis of CGH findings pointed to the chromosomal region 13q21-q22 as the site containing a novel breast cancer predisposition locus. The presence of this locus was supported by linkage analysis of 77 Finnish, Icelandic, and Swedish breast cancer pedigrees, which provided suggestive evidence of a breast cancer susceptibility locus at 13q21-q22. Because of the substantial genetic heterogeneity of breast cancer, studies to evaluate the significance of this candidate locus in other populations will be critically important.
Acknowledgments
We thank Kati Rouhento, Kristiina Selkee, Bjarnveig I Sigurbjörnsdottir, Sigrun Kristjansdottir, Kristrun Olafsdottir, Omar Kristinsson, Anna Gudrun Hafsteinsdottir, Oddny Vilhjalmsdottir, Hrafn Tulinius, Gudridur Olafsdottir, Natalie Goldberger, Minna Merikivi, and the Finnish Cancer Registry for assistance and information. The study was supported by the National Institutes of Health, Nordic Cancer Society, Finnish Cancer Society, Academy of Finland, Sigrid Juselius Foundation, Clinical Research Funds of the Tampere and Helsinki University Hospitals, Icelandic Research Council, The Icelandic University Hospital Research Fund, Swedish Cancer Society, Mrs. Berta Kamprads Foundation, the G.A.E. Nilsson Foundation, the F&M Bergqvist Foundation, the King Gustav V:s Jubilee Foundation, the Finnish Cultural Foundation, Emil Aaltonen Foundation, and the Maud Kuistila Foundation.
Abbreviations
- CGH
comparative genomic hybridization
- lod
logarithm of odds
- Hlod
heterogeneity lod score
References
- 1.Ford D, Easton D F, Stratton M, Narod S, Goldgar D, Devilee P, Bishop D T, Weber B, Lenoir G, Chang-Claude J, et al. Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vehmanen P, Friedman L S, Eerola H, McClure M, Ward B, Sarantaus L, Kainu T, Syrjakoski K, Pyrhonen S, Kallioniemi O-P, et al. Hum Mol Genet. 1997;6:2309–2315. doi: 10.1093/hmg/6.13.2309. [DOI] [PubMed] [Google Scholar]
- 3.Håkansson S, Johannsson O, Johansson U, Sellberg G, Loman N, Gerdes A M, Holmberg E, Dahl N, Pandis N, Kristoffersson U, et al. Am J Hum Genet. 1997;60:1068–1078. [PMC free article] [PubMed] [Google Scholar]
- 4.Knudson A G. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemminki A, Tomlinson I, Markie D, Jarvinen H, Sistonen P, Bjorkqvist A M, Knuutila S, Salovaara R, Bodmer W, Shibata D, et al. Nat Genet. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- 6.Mehenni H, Blouin J L, Radhakrishna U, Bhardwaj S S, Bhardwaj K, Dixit V B, Richards K F, Bermejo-Fenoll A, Leal A S, Raval R C, et al. Am J Hum Genet. 1997;61:1327–1334. doi: 10.1086/301644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tirkkonen M, Johannsson O, Agnarsson B A, Olsson H, Ingvarsson S, Karhu R, Tanner M, Isola J, Barkardottir R B, Borg Å, et al. Cancer Res. 1997;57:1222–1227. [PubMed] [Google Scholar]
- 8.Desper R, Jiang F, Kallioniemi O-P, Moch H, Papadimitriou C H, Schäffer A A. J Comput Biol. 1999;6:37–51. doi: 10.1089/cmb.1999.6.37. [DOI] [PubMed] [Google Scholar]
- 9.Desper, R., Jiang, F., Kallioniemi, O.-P., Moch, H., Papadimitriou, C. H. & Schäffer, A. A. (2000) J. Comput. Biol., in press. [DOI] [PubMed]
- 10.Tirkkonen M, Tanner M, Karhu R, Kallioniemi A, Isola J, Kallioniemi O-P. Genes Chromosomes Cancer. 1998;21:177–184. [PubMed] [Google Scholar]
- 11.Kuukasjärvi T, Tanner M, Pennanen S, Karhu R, Visakorpi T, Isola J. Genes Chromosomes Cancer. 1997;18:94–101. [PubMed] [Google Scholar]
- 12.Brodeur G M, Tsiatis A A, Williams D L, Luthardt F W, Green A A. Cancer Genet Cytogenet. 1982;2:137–152. doi: 10.1016/0165-4608(82)90010-3. [DOI] [PubMed] [Google Scholar]
- 13.Morton N E. Proc Natl Acad Sci USA. 1991;88:7474–7476. doi: 10.1073/pnas.88.17.7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu Y J, Liu T H. Sci Sinica. 1965;14:1396–1400. [Google Scholar]
- 15.Edmonds J. J Res Nat Bur Stand. 1967;71:233–240. [Google Scholar]
- 16.Karp R M. Networks. 1971;1:265–272. [Google Scholar]
- 17.Tarjan R E. Networks. 1977;7:25–35. [Google Scholar]
- 18.Feltenstein J. Cladistics. 1989;5:164–166. [Google Scholar]
- 19.Smith J R, Freije D, Carpten J D, Gronberg H, Xu J, Isaacs S D, Brownstein M J, Bova G S, Guo H, Bujnovszky P, et al. Science. 1996;274:1371–1374. doi: 10.1126/science.274.5291.1371. [DOI] [PubMed] [Google Scholar]
- 20.O'Connell J R, Weeks D E. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott J. Proc Natl Acad Sci USA. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lathrop G M, Lalouel J M, Julier C, Ott J. Proc Natl Acad Sci USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cottingham R W, Jr, Idury R M, Schäffer A A. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 24.Schäffer A A, Gupta S K, Shriram K, Cottingham R W., Jr Hum Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 25.Claus E B, Risch N, Thompson W D. Am J Hum Genet. 1991;48:232–242. [PMC free article] [PubMed] [Google Scholar]
- 26.Easton D F, Bishop D T, Ford D, Crockford G P. Am J Hum Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- 27.Ott J. Analysis of Human Genetic Linkage. Baltimore: Johns Hopkins Univ. Press; 1992. [Google Scholar]
- 28.Kruglyak L, Daly M J, Reeve-Daly M P, Lander E S. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 29.Weeks D E, Ott J, Lathrop G M. Am J Hum Genet. 1990;47:A204. (abstr.). [Google Scholar]
- 30.Eiriksdottir G, Johannesdottir G, Ingvarsson S, Bjornsdottir I B, Jonasson J G, Agnarsson B A, Hallgrimsson J, Gudmundsson J, Egilsson V, Sigurdsson H, et al. Eur J Cancer. 1998;34:2076–2081. doi: 10.1016/s0959-8049(98)00241-x. [DOI] [PubMed] [Google Scholar]
- 31.Hyytinen E R, Frierson H F, Jr, Boyd J C, Chung L W, Dong J T. Genes Chromosomes Cancer. 1999;25:108–114. [PubMed] [Google Scholar]
- 32.Yin Z, Spitz M R, Babaian R J, Strom S S, Troncoso P, Kagan J. Oncogene. 1999;18:7576–7583. doi: 10.1038/sj.onc.1203203. [DOI] [PubMed] [Google Scholar]
- 33.Hu N, Roth M J, Emmert-Buck M R, Tang Z Z, Polymeropolous M, Wang Q H, Goldstein A M, Han X Y, Dawsey S M, Ding T, et al. Clin Cancer Res. 1999;5:3476–3482. [PubMed] [Google Scholar]
- 34.Tsang Y S, Lo K W, Leung S F, Choi P H, Fong Y, Lee J C, Huang D P. Int J Cancer. 1999;83:305–308. doi: 10.1002/(sici)1097-0215(19991029)83:3<305::aid-ijc3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 35.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al. Nature (London) 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 36.Jenne D E, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Nat Genet. 1998;1:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Silver D P, Walpita D, Cantor S B, Gazdar A F, Tomlinson G, Couch F J, Weber B L, Ashley T, Livingston D M, et al. Mol Cell. 1998;2:317–338. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]