

Abstract
Overexpression of ErbB-2/Neu has been causally associated with mammary epithelial transformation. Here we report that blockade of the epidermal growth factor receptor (EGFR) kinase with AG-1478 markedly delays breast tumor formation in mouse mammary tumor virus (MMTV)/Neu + MMTV/transforming growth factor α bigenic mice. This delay was associated with inhibition of EGFR and Neu signaling, reduction of cyclin-dependent kinase 2 (Cdk2) and mitogen-activated protein kinase (MAPK) activities and cyclin D1, and an increase in the levels of the Cdk inhibitor p27Kip1. In addition, BrdUrd incorporation into tumor cell nuclei was prevented with no signs of tumor cell apoptosis. These observations prompted us to investigate the stability of p27. Recombinant p27 was degraded rapidly in vitro by untreated but not by AG-1478-treated tumor lysates. Proteasome depletion of the tumor lysates, addition of the specific MEK1/2 inhibitor U-0126, or a T187A mutation in recombinant p27 all prevented p27 degradation. Cdk2 and MAPK precipitates from untreated tumor lysates phosphorylated recombinant wild-type p27 but not the T187A mutant in vitro. Cdk2 and MAPK precipitates from AG-1478-treated tumors were unable to phosphorylate p27 in vitro. These data suggest that increased signaling by ErbB receptors up-regulates MAPK activity, which, in turn, phosphorylates and destabilizes p27, thus contributing to dysregulated cell cycle progression.
The ErbB family of receptors includes the epidermal growth factor receptor (EGFR) ErbB-1, the orphan ErbB-2 (Neu), and the Neuregulin receptors ErbB-3 and ErbB-4 (1–4). Binding of ligands to the ectodomain of these receptors results in the formation of homodimeric and heterodimeric complexes (5), which is followed rapidly by the activation of the receptors' intrinsic tyrosine kinase. Consequently, phosphorylation of specific C-terminal tyrosine residues and the recruitment of specific second messengers activate a plethora of intracellular signaling pathways that play central roles in cell proliferation, development, differentiation, migration, and oncogenesis (6).
Many studies support a pivotal role for the orphan ErbB-2/Neu in ErbB signaling and tumorigenesis (7–9), suggesting that this receptor plays a critical role in the cellular responses mediated by the ligand-dependent activation of other ErbB receptors (10, 11). Indeed, overexpression of ErbB-2/Neu alone or in combination with EGFR (ErbB-1) or ErbB-3 in vitro can transform mammary epithelial cells (8) and fibroblasts (12, 13), respectively. A central role for ErbB-2/Neu in transformation has been shown by using transgenic mice overexpressing the protooncogene under the control of the mouse mammary tumor virus (MMTV) promoter (14, 15). In these tumors, DNA sequence analysis revealed the presence of a 16-aa in-frame deletion in the extracellular domain of ErbB-2/Neu, resulting in a constitutively activated receptor capable of transforming Rat-1 fibroblasts (16). So far, no such activating mutations have been found in human tumors, where the most common change involving the protooncogene is mRNA and protein overexpression with or without gene amplification. Nonetheless, clinical surveys have shown overexpression of nonmutant ErbB-2/Neu in a subset of epithelial neoplasms with a particularly virulent behavior (17, 18). Moreover, antibodies against the ectodomain of ErbB-2/Neu can alter the natural history of breast carcinomas that overexpress the protooncogene (19). These observations provide evidence for a critical role of the ErbB-2/Neu receptor protein in mammary transformation and tumor progression.
The lack of activating ErbB-2/Neu mutations in human breast tumors suggests that this receptor can be transactivated through ligand-activated ErbB coreceptors present in the tumor cells. Consistent with this paradigm, we reported a synergistic interaction between MMTV/Neu and MMTV/transforming growth factor α (TGF-α), one of the EGFR ligands, in the induction of mammary tumors in virgin female transgenic mice (20). The mammary glands in these mice display a range of sequential histological changes including extensive lobuloalveolar development, epithelial hyperplasia and dysplasia, carcinoma in situ, invasive cancers, and distant metastases, reminiscent of the progression observed in human breast cancer. Tumors arising in these bigenic mice contain tyrosine-phosphorylated wild-type ErbB-2/Neu (20), supporting ErbB-2/Neu transactivation by the TGF-α-stimulated EGFR kinase.
In this model of receptor cooperativity, we have tested the effect of a quinazoline small-molecule inhibitor of the EGFR tyrosine kinase, AG-1478, on Neu/TGF-α-mediated mammary transformation. Although relatively EGFR-specific at low concentrations, we have shown that this inhibitor can induce inactive, unphosphorylated EGFR–ErbB-2/Neu heterodimers, thereby sequestering ErbB-2/Neu from signaling interactions with other ErbB coreceptors (21). We now report that AG-1478 inhibits EGFR as well as ErbB-2/Neu signaling and alters the natural history of MMTV/Neu + TGF-α breast tumors most likely by modulating downstream kinases that regulate molecules involved in G1/S traverse, thereby inducing cell cycle arrest.
Materials and Methods
Cell Lines, Kinase Inhibitors, and Antibodies.
MCF-10A/TE cells, transfected with ErbB-2/Neu and TGF-α (22), were provided by D. Salomon (National Institutes of Health, Bethesda, MD) and maintained in DMEM/Ham's F-12 medium supplemented with 5% FCS (Atlanta Biologicals, Norcross, GA), 10 μg/ml insulin, and 0.5 μg/ml hydrocortisone (Sigma). SKBR-3 and BT-474 human breast cancer cells were from the American Type Culture Collection and were grown in improved MEM (IMEM)/10% FCS. All cells were maintained at 37°C in 5% CO2. AG-1478, an EGFR kinase inhibitor with an IC50 of 3 nM in vitro (21), was prepared as a 10-mM stock solution in DMSO (Sigma). U-0126, a specific inhibitor of MEK1 and MEK2 (23), was from Calbiochem. For immunoblot analysis, the following antibodies were used: EGFR, Shc, p27, and PLC-γ1 (Transduction Laboratories, Lexington, KY); ErbB-2/Neu (NeoMarkers, Fremont, CA); phosphotyrosine (P-Tyr; Upstate Biotechnology, Lake Placid, NY); cyclin D1 and Rb (PharMingen); Grb-2 and cyclin A (Santa Cruz Biotechnology); active MAP kinase (Promega); and p21Cip1 (Oncogene Science).
Cell Proliferation Assays and Flow Cytometric Analysis.
MCF-10A/TE cells were sparsely plated in 6-well plates in regular growth medium with or without 10 μM AG-1478. After 5 days, the monolayers were trypsinized and cell numbers were determined in a Coulter counter. The effect of AG-1478 against BT-474 and SKBR-3 tumor cells was tested in a soft agarose colony-forming assay. Cells were plated at a density of 3 × 104 cells per 35-mm dish in IMEM/10% FCS/0.8% agarose/10 mM Hepes with or without 1–10 μM AG-1478 as described (24). After a 7-day incubation at 37°C, colonies measuring ≥50 μm were counted by using an Omnicon FAS III image analyzer (Bausch & Lomb). Flow cytometric analysis of propidium iodide-labeled cell nuclei was performed as described (25).
Cell Lysis and Immunoblot Analyses.
MCF-10A/TE, BT-474, and SKBR-3 cells were treated with 0.5–10 μM AG-1478 for 24 h and then lysed for 20 min at 4°C in EBC buffer as described (21). Total protein (75 μg) was resolved by SDS/PAGE, transferred to nitrocellulose, and subjected to immunoblot analyses with antibodies against EGF-R, Neu, P-Tyr, cyclin D1, Rb, and p27 (see above). Blots were incubated with horseradish peroxidase-linked IgG secondary antibodies (Amersham Pharmacia). Immunoreactive bands were detected by chemiluminescence (Roche Molecular Biochemicals).
Studies in Bigenic Mammary Tumors.
MMTV/Neu + TGF-α bigenic mice were generated as described (15, 26). At 8 weeks, 10 mice per group were randomized to either daily i.p injections with 50 mg/kg AG-1478 or DMSO alone and evaluated daily thereafter for the occurrence of mammary tumors. Mammary gland tissue was harvested after 6 months from both control and mice treated with AG-1478. Samples were fixed overnight in Histoprep (Fisher Scientific), sectioned, and stained with hematoxylin/eosin. In other cases, whole gland-mount preparations were performed as described (27). To evaluate the antiproliferative effect of AG-1478 in situ, bigenic mice with and without palpable tumors were treated for 5 consecutive days with the EGFR inhibitor or DMSO. On the fifth day, BrdUrd (50 mg/kg in PBS) was administered i.p. 2 h before tissue excision. Tissue sections were incubated with a BrdUrd antibody (Biogenics), and BrdUrd-labeled nuclei were visualized by using the Histo-mouse Kit (Zymed). Additional sections were used to stain for apoptotic cells by using the terminal deoxynucleotidyltransferase-mediated UTP-end-labeling method (Intergen, Purchase, NY). For evaluation of tumor microvessels, sections were stained with a polyclonal antibody against factor VIII (Dako).
Pharmacokinetic Studies.
Plasma analyses of AG-1478 were performed in 20-gm female BALB/c nu/nu mice. AG-1478 (50 mg/kg) was administered i.p. in 100 μl of DMSO. Blood samples were collected at 0, 5, 15, 30, and 45 min and 1, 3, and 6 h postdose (four mice per time point) by cardiac puncture. Plasma was prepared by centrifugation (14,000 rpm, 10 min, 4°C) and stored at −80°C. For extraction of AG-1478 from plasma, an internal standard was added to 200-μl plasma aliquots. Samples were extracted by addition of 3 × 0.5 ml of acetonitrile followed by vortexing (high speed) for approximately 1 min. Precipitated protein was removed by centrifugation. The supernatants were collected and evaporated to dryness in a Speed-Vac apparatus. Residues were reconstituted with 100 μl of methanol/mobile phase (50:50, vol/vol). Calibration standards of AG-1478 were prepared in an identical manner by using heat-inactivated pooled plasma. Samples were analyzed by HPLC using a Hypersil (4.6 × 100 mm, 5-μM particle size) column and using a mobile phase consisting of tetrahydrofuran/0.02 M KH2PO4/triethylamine (42.85:57:0.15, vol/vol/vol) with a 10-μl injection, a flow rate (isocratic) of 1.0 ml/min, and detection at 325 nm. The lower limit of detection was 0.5 μg/ml (1.5 μM).
Immunoprecipitation and Immunoblot Analysis.
Mammary glands or tumors were isolated before and after five daily injections with AG-1478 and then homogenized by using a Polytron Brinkmann homogenizer in TNE lysis buffer (50 mM Tris⋅HCl, pH 7.6/150 mM NaCl/2 mM EDTA/1 mM Na3VO4/2 mM DTT/10 μg/ml aprotinin and leupeptin). Nonidet P-40 (1%, vol/vol) was added, and the samples were incubated for 20 min on ice. Lysates were cleared by centrifugation (12,000 rpm, 10 min, 4°C), and 3 mg of total protein was immunoprecipitated overnight at 4°C with either 300 ng of an EGFR (Santa Cruz Biotechnology) or an ErbB-2/Neu (Neomarkers) polyclonal antibody, each followed by protein A-Sepharose (Sigma) for 2 h at 4°C. Precipitates were resolved by SDS/PAGE and then subjected to EGFR, ErbB-2/Neu, P-Tyr, PLC-γ1, Shc, or Grb-2 immunoblot analyses. In other cases, 100 μg of total protein was subjected to immunoblot procedures for active MAP kinase, Rb, p27, p21, cyclin D1, and cyclin A.
p27Kip1 Degradation Assay.
Approximately 10 mm3 of snap-frozen mouse mammary tumor tissue was minced in 100 μl of ice-cold degradation buffer (20 mM Tris⋅HCl, pH 7.6/2 mM DTT/0.25 mM EDTA/10 μg/ml leupeptin/pepstatin). Lysates were freeze-thawed three times in liquid N2 and cleared by centrifugation at 13,000 rpm at 4°C for 10 min. Proteasome-dependent p27 degradation activity was determined by incubating 250 ng of recombinant histidine-tagged wild-type p27 or mutant p27 (Thr-187 replaced by Ala; kindly provided by M. Pagano, New York University) with 20 μg of total tumor protein at 30°C (28). Where indicated, the proteasome complex was eliminated from tumor lysates by differential centrifugation at 100,000 × g for 30 min at 4°C as described (28). After incubation at 30°C for times ranging from 30 min to 20 h, the samples were subjected to p27 immunoblot analysis.
In Vitro Kinase Assays.
Cyclin-dependent kinase 2 (Cdk2), MEK1, or total MAPK was precipitated overnight at 4°C from 1 mg of tumor lysate with Cdk2 (Santa Cruz Biotechnology) or MAPK (New England Biolabs) polyclonal antibodies or with a mAb against MEK1 (Transduction Laboratories), each with protein A-Sepharose CL-4B (Sigma). After washes with ice-cold PBS (four times) and specific kinase buffer, the immune complexes were resuspended in 30 μl of kinase assay buffer (Cdk2 kinase buffer: 50 mM Hepes, pH 7.5/10 mM MgCl2/1 mM DTT/2.5 mM EGTA/0.1 mM Na3VO4/1 mM ATP; MAPK kinase buffer: 30 mM Hepes, pH 7.4/15 μM ATP/15 mM MgCl2; MEK kinase buffer: 20 mM Hepes, pH 7.0/5 mM 2-mercaptoethanol/10 mM MgCl2/0.1 mM Na3VO4/10 μM ATP) containing either 1 μg of histone H1 (HH1; Boehringer Mannheim), 5 μg of myelin basic protein (MBP; Sigma), 5 μg of glutathione S-transferase-MAPK (Upstate Biotechnology), 10 μg of wild-type p27-His, or T187A p27-His. [γ-32P]ATP (10 μCi; specific activity, 3,000 Ci/mmol; Amersham Pharmacia) was added to each reaction, and the samples were incubated for 30 min at 30°C (for Cdk2 and MAPK) or for 25 min at 25°C (for MEK1). Kinase reactions were terminated by adding Laemmli sample buffer followed by boiling for 5 min. Phosphorylated substrates were resolved by SDS/PAGE and visualized by autoradiography.
Results
EGFR Kinase Blockade Inhibits the Proliferation of Neu-Overexpressing Cells.
A 24-h incubation of MCF-10A/TE cells with AG-1478 inhibited basal tyrosine phosphorylation of a 180-kDa band, most likely representing EGFR and ErbB-2/Neu, in a dose-dependent manner (Fig. 1a). A similar effect was observed in BT-474 and SKBR-3 breast tumor cells, which exhibit ErbB-2/Neu gene amplification and approximately 1–2 × 105 EGF-binding sites per cell. These occurred without any change on EGFR or ErbB-2/Neu protein levels (Fig. 1 a and c) but was associated with an inhibitory effect on basal cell proliferation. Five days of continuous or intermittent, 1-h daily treatment with 10 μM AG-1478 inhibited proliferation of MCF-10A/TE cell monolayers by 70% and 40%, respectively (Fig. 1b). Colony formation of both BT-474 and SKBR-3 cells was reduced in a dose-dependent manner by 1–10 μM AG-1478 with no detectable colony formation at the higher concentration (Fig. 1d). In both tumor lines, this was accompanied by a reduction in phosphorylated Rb (p-Rb) and cyclin D1 levels and an increase in the Cdk inhibitor p27 (Fig. 1c), suggesting that blockade of EGFR and ErbB-2/Neu signaling resulted in inhibition of cell cycle progression. Cell cycle arrest was confirmed by flow cytometric analysis. A 24-h incubation with 10 μM AG-1478 resulted in G1 arrest and a marked reduction of the S phase fraction in both BT-474 and SKBR-3 cells (Fig. 1e). These effects were reversible upon removal of AG-1478 with no evidence of cell death under these conditions.
Figure 1.

Blockade of the EGFR kinase inhibits the proliferation of ErbB-2/Neu-overexpressing cells. (a) MCF-10A/TE cells with or without AG-1478. After 24 h, cell lysates were prepared in EBC buffer and subjected to immunoblot procedures for EGFR, ErbB-2/Neu, and P-Tyr. Levels of a 180-kDa P-Tyr band, probably representing EGFR and ErbB-2/Neu, were reduced by AG-1478 without any change in receptor protein content. (b) MCF-10A/TE cells were seeded in 6-well plates at a density of 0.5 × 106 cells per well and treated either continuously for 5 days or daily for 1 h for 5 consecutive days with 10 μM AG-1478. Each bar represents the mean number of cells ± SE of three wells after the 5-day experiment. The hatched bar represents the number of cells per well 24 h postplating. (c) Exponentially growing BT-474 and SKBR-3 breast tumor cells were incubated overnight with or without 10 μM AG-1478, solubilized in EBC buffer, and then analyzed by immunoblot procedures for ErbB-2/Neu, P-Tyr, Rb, cyclin D1, and p27. (d) BT-474 and SKBR-3 cells (3 × 104) were plated in 35-mm dishes in IMEM/10% FCS/0.8% agarose/10 mM Hepes in the absence or presence of 1–10 μM AG-1478. After 7 days, colonies measuring 50 μm were counted. Each bar represents the mean colony number ± SE of three dishes. (e) BT-474 and SKBR-3 cells were incubated overnight with 10 μM AG-1478 and then subjected to flow cytometric analysis. Numbers represent the percentage of cells in G1, G2M, and S phases.
EGFR Kinase Blockade Suppresses Mammary Tumorigenicity in MMTV/Neu + TGF-α Mice.
Recent studies have shown that 50 mg/kg per day of AG-1478 given i.p. to nude mice can markedly delay tumor growth of NIH 3T3 tumors transfected with ectopic ErbB-2/Neu (L.K.S., unpublished data). In addition, Muller et al. (20) showed that 50% of MMTV/Neu + TGF-α bigenic mice develop spontaneous mammary tumors by 24 weeks. Therefore, we examined the effect of EGFR kinase blockade with AG-1478 on breast tumor development in this mouse transgenic model. Eight-week-old virgin female mice were allocated randomly to either daily i.p. injections with 50 mg/kg AG-1478 or DMSO (control). At 28 weeks of age, 50% of the control mice had developed at least one palpable breast tumor, whereas all mice treated with AG-1478 remained tumor-free. AG-1478 treatment was discontinued at 36 weeks of age, at which time 9 of 10 control mice and 2 of 10 experimental mice exhibited at least one palpable tumor (Fig. 2a). Histologically, all tumors were invasive or in situ ductal carcinomas. Whole-mount gland preparations (Fig. 2 b and c) and hematoxylin/eosin-stained histological sections from mammary glands without a palpable tumor (Fig. 2 d and e) revealed extensive lobuloalveolar development and epithelial hyperplasia and dysplasia with occasional foci of carcinoma in situ in the controls, whereas the glands from AG-1478-treated mice displayed a normal to hypoplastic ductal tree. No signs of toxicity were observed. Two months after cessation of treatment, 7 of 10 (70%) of the AG-1478-treated mice developed mammary tumors, suggesting that the cytostatic effect of AG-1478 depended on the continuous presence of the quinazoline.
Figure 2.

Blockade of the EGFR kinase prolongs the latency of MMTV/Neu + TGF-α bigenic mammary tumors. (a) Eight-week-old bigenic mice were allocated randomly to daily i.p. injections with 50 mg/kg AG-1478 (○) or DMSO (▴). After 28 weeks, 9 of 10 (90%) mice in the control group developed at least one mammary tumor, whereas only 2 of 10 (20%) of the AG-1478-treated mice developed palpable tumors (P = 0.0007 on day 250; P = 0.006 on day 300 by log-rank test). (b and c) Whole-mount preparations from control and 6-month-old bigenic mice treated with DMSO (b) or AG1478 (c). (d and e) Hematoxylin/eosin-stained ×400 histological sections of mammary glands from 6-month-old bigenic mice treated with DMSO (d) or AG-1478 (e).
Pharmacokinetic studies revealed that 50 mg/kg i.p. of AG-1478 resulted in a peak plasma concentration of 19.3 μg/ml (61 μM) with a half-life of 31 min. Plasma levels >1.5 μM, above the concentrations known to inhibit EGFR kinase function, were observed for longer than 3 h after a single 50-mg/kg i.p. injection. Although it is not known what the levels of AG-1478 in the mammary gland are, the lipophillicity of the compound and high volume of distribution (>3 liters/kg) support the ability of AG-1478 to penetrate tissues and cells.
To determine whether AG-1478 was antiproliferative or cytotoxic, we treated 4-month-old tumor-free mice with AG-1478 for 5 consecutive days immediately followed by a BrdUrd pulse. Before treatment, mammary epithelium exhibited 10 BrdUrd-labeled nuclei per 100 cells, whereas these were undetectable in glands from mice treated with AG-1478 (Fig. 3). This indicated a remarkable inhibition of DNA synthesis in situ as a result of EGFR blockade. Similarly, treatment of mice with established carcinomas with a similar 5-day schedule reduced the number of mitotic counts by 50% compared with those measured before therapy (not shown). To determine whether programmed cell death was occurring in vivo, apoptosis was determined in the same tissue sections by terminal deoxynucleotidyltransferase-mediated UTP end labeling assay. Post-AG-1478 mammary glands did not display any apoptotic nuclei. However, apoptotic nuclei were detectable in untreated transgenic tumors and 24 h after a single i.p. dose of 25 mg/kg of the anticancer drug cisplatin (data not shown).
Figure 3.
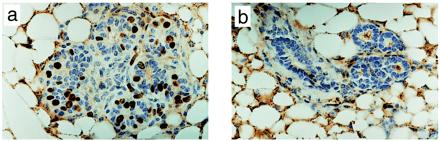
AG-1478 inhibits DNA synthesis in mammary epithelium from MMTV/Neu + TGF-α bigenic mice. (a and b) Mice were injected daily for 5 days with DMSO (a) or AG-1478 (b). On the fifth day, BrdUrd was injected i.p. 2 h before isolation of one of the inguinal mammary glands. Tissue sections were prepared and subjected to immunohistochemistry with a BrdUrd antibody as described in Materials and Methods. (Magnification: ×400.)
Blockade of EGFR/Neu Signaling Alters the Content of Molecules Involved in G1/S Traverse.
Two mice with multiple tumors were selected to compare the biochemical targets of AG-1478 in different tumors within the same mouse before and after treatment. A 5-day treatment with 50 mg/kg AG-1478 reduced the P-Tyr content of both EGFR and ErbB-2/Neu without reducing receptor protein levels (Fig. 4a). As a consequence, the association of both EGFR and ErbB-2/Neu with Shc and PLC-γ1 but less with Grb-2, second messengers that interact with phosphotyrosines in the receptors' C terminus, was reduced by AG-1478 (Fig. 4a). In addition, constitutively active MAP kinase, as measured with an antibody against phospho-MAPK p42/p44, was eliminated by treatment with the kinase inhibitor (Fig. 4b). Because of the previously reported effects of MAP kinase on both the transcription of cyclin D1 and cyclin A (29) as well as on the down-regulation of the Cdk inhibitor p27 (30, 31), we also measured the content of these proteins. Consistent with the inhibition of BrdUrd incorporation, treatment with AG-1478 reduced the levels of cyclin D1 and p-Rb while up-regulating p27 levels. The levels of cyclin A and the Cdk inhibitor p21 were reduced modestly by AG-1478 (Fig. 4b).
Figure 4.

AG-1478 treatment blocks EGFR and ErbB-2/Neu signaling and modulates molecules involved in G1/S traverse. (a) EGFR and ErbB-2/Neu proteins were precipitated from tumor tissue taken from two mice (I, II) immediately before (−) and after (+) five i.p. doses of AG-1478 (50 mg/kg per day). The immune complexes were subjected to immunoblot procedures by using EGFR, ErbB-2/Neu, P-Tyr, Shc, Grb-2, and PLC-γ1 antibodies. (b) Protein (100 μg/lane) from the mammary tumor lysates from the same mice before (−) and after (+) treatment was subjected to immunoblot analyses with antibodies against active MAPK, Rb, cyclin D1, cyclin A, p27, and p21.
Blockade of the EGFR and ErbB-2/Neu Signaling Down-Regulates Cdk2 Activity and Stabilizes p27.
The complete elimination of BrdUrd labeling and the loss of p-Rb suggested that AG-1478 inhibited tumor Cdk2 activity. Cyclin E/Cdk2-mediated phosphorylation of p27 on Thr-187 results in proteasome-mediated degradation of p27 and entry into S of mouse fibroblasts (32). Therefore, we examined both Cdk2 activity and stability of recombinant p27 in transgenic tumor lysates. Cdk2 precipitates from a tumor lysate phosphorylated both HH1 and wild-type p27-His but not a T187A p27 mutant in vitro. Cdk2 activity against HH1 and p27 was reduced markedly in tumors from mice treated with AG-1478 (Fig. 5a). If activated EGFR and/or ErbB-2/Neu signaling contributes to proteasome-mediated degradation of p27, then treatment with AG-1478 in vivo, perhaps via inhibition of Cdk2 activity (Fig. 5a), or proteasome depletion in vitro should prevent p27 degradation. Indeed, recombinant p27 was degraded within 30 min at 30°C by a tumor lysate from a DMSO-treated (control) mouse whereas it was completely stable for up to 20 h when no tumor lysate was added. This degradation was abolished by depletion of the proteasome complex. In addition, recombinant p27 remained intact for >3 h when incubated with a tumor lysate obtained from a mouse treated with 50 mg/kg AG-1478 (Fig. 5b), agreeing with the induced increase in the steady-state levels of p27 (Fig. 4B). A mutant T187A p27 was resistant to tumor lysate-induced degradation in vitro, implying that the latter required phosphorylation on T187 (Fig. 5c).
Figure 5.
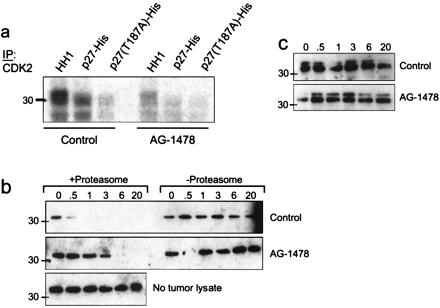
Phosphorylation/degradation of recombinant p27 by MMTV/Neu + TGF-α tumor lysates. (a) Cdk2 was precipitated from tumor lysates from mice that had been treated or not with AG-1478 and added to a kinase reaction containing either HH1, wild-type p27, or T187A p27 as substrates for [γ-32P]ATP incorporation. Phosphorylated products were resolved by SDS/PAGE and visualized by autoradiography. (b and c) Twenty micrograms of total protein from the tumor lysates was incubated with 250 ng of either wild-type p27 (b) or T187A p27 (c) at 30°C for the indicated times. Where indicated, tumor lysates were depleted of their proteasome complex by differential centrifugation before incubation with p27. To monitor for spontaneous degradation, wild-type p27-His was incubated under the same conditions but in the absence of a tumor lysate (b Bottom). Degradation was assessed by SDS/PAGE and p27 immunoblot as indicated in Materials and Methods. The endogenous p27 is undetectable by immunoblot analysis of this amount of total protein (20 μg) from the tumor lysates. Because of the histidine tag, both wild-type and mutant p27 molecules migrate as 33-kDa proteins.
Tumor MAPK Activity Phosphorylates and Destabilizes p27.
It has been reported that activation of ras/MAPK contributes to cell cycle progression by posttranslationally modifying and destabilizing p27 (30, 31). Because blockade of EGFR and ErbB-2/Neu signaling eliminated active MAPK and increased p27 levels, we examined the contribution of tumor MAPK to the degradation of p27 by using a MEK1/2 inhibitor. In vitro, U-0126 inhibited tumor MEK1 activity against glutathione S-transferase-MAPK and tumor MAPK activity against MBP but not tumor Cdk2 activity (Fig. 6a). Consistent with the elimination of phospho-MAPK in AG-1478-treated tumors (Fig. 4b), MEK1 and MAPK in vitro activities were reduced markedly in a tumor from an AG-1478-treated mouse compared with a control tumor (Fig. 6a). To test whether activated EGFR and ErbB-2/Neu mediated the degradation of p27 via MAPK, we tested the stability of recombinant p27 when incubated with a tumor lysate in the presence of the MEK1/2 inhibitor. In the presence of U-0126, recombinant p27 was stable for up 20 h, whereas, in its absence, complete degradation is achieved after 1 h. Similar to Cdk2 (Fig. 6b), MAPK precipitated from a control tumor phosphorylated wild-type p27 but not T187A p27 (Fig. 6c), implying that activated MAPK phosphorylates p27 on T187, thus contributing to degradation of the Cdk inhibitor in the transgenic tumors.
Figure 6.
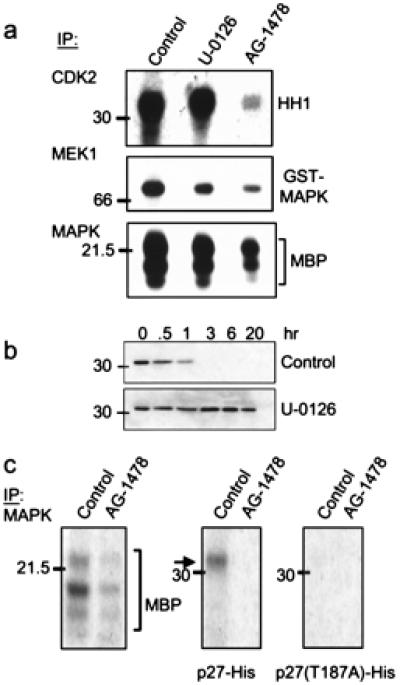
MAPK-mediated phosphorylation/degradation of p27. (a) Cdk2, MEK1, and MAPK were precipitated from a tumor lysate from a treatment-naïve mouse (control and U-0126 lanes) or from a mouse that had been treated with AG-1478 for 5 days. Immune complexes then were added to kinase reactions containing HH1, glutathione S-transferase-MAPK, or MBP, respectively. Where indicated, 10 μM U-0126 was added to the kinase reaction. Phosphorylated species were resolved by SDS/PAGE and visualized by autoradiography. (b) Twenty micrograms of total protein from a proteasome-containing tumor lysate was incubated with 250 ng of p27-His at 30°C for the indicated times in the presence of 10 μM U-0126 or DMSO (control). Degradation of p27 was monitored by immunoblot analysis as described in Fig. 5. (c) MAPK was precipitated from tumor lysates from mice that had been treated or not with AG-1478. Immune complexes then were tested for kinase activity against MBP, p27-His, or T187A p27 substrates. After SDS/PAGE, phosphorylated substrates were visualized by autoradiography.
Discussion
We have examined the effect of EGFR tyrosine kinase blockade on the natural history of MMTV/Neu + TGF-α transgenic tumors. Daily systemic administration of AG-1478, a small-molecule EGFR kinase inhibitor, almost completely prevented tumor formation. Although AG-1478 is specific for the EGFR at submicromolar concentrations (21, 33), several arguments support its use to block EGFR–ErbB-2/Neu crosstalk and signaling. AG-1478 has been shown to stabilize inactive, unphosphorylated EGFR–ErbB-2/Neu heterodimers. This inactive heterodimerization stabilized by AG-1478 abrogates heregulin-mediated signaling in ErbB-2/Neu-overexpressing SKBR-3 human breast cancer cells (21), perhaps by preventing ErbB-2/Neu from interacting with other ErbB coreceptors. Even if ErbB-2/Neu was not a direct target of AG-1478, we should note that, in this model, it is the EGFR kinase that predominantly transactivates the ErbB-2/Neu kinase after autocrine/paracrine stimulation by the TGF-α transgene (20). Even though the in vivo half-life of AG-1478 is 31 min, this concentration is logarithmically higher than that required to inhibit the EGFR kinase as well as intact cells with autoactivated EGFR (21, 33). Furthermore, plasma concentrations >1.5 μM were observed 3 h after administration of a single, 50-mg/kg dose of AG-1478. That the lower limit of detection of AG-1478 in plasma by HPLC was 1.5 μM, well in excess of those that inhibit EGFR function, further suggests that effective EGFR kinase-inhibiting levels may have been more prolonged than what was estimated. Moreover, the lipophillicity and high volume of distribution of AG-1478 indicated by the pharmacokinetic studies in nude mice suggest adequate penetration in mammary tissues.
A brief, 5-day treatment with AG-1478 of tumor-free and tumor-bearing mice revealed that this treatment resulted in the inhibition of EGFR and ErbB-2/Neu tyrosine phosphorylation as well as elimination or inhibition of the association of these receptors with several signal-transducing molecules. The antitumor effect appeared to be predominantly cytostatic, based on the following observations: (i) most mice developed mammary tumors upon interruption of treatment; (ii) there was no evidence of apoptosis in situ but an almost complete elimination of BrdUrd incorporation into epithelial cell nuclei; and (iii) there were changes in content of molecules involved in the G1-to-S transition with elimination of Cdk2 activity and p-Rb levels. Recent studies suggest that perturbation of EGFR and/or ErbB-2/Neu in receptor-overexpressing tumor systems may result in an antitumor effect, at least in part, via the interruption of tumor–host interactions that are critical for neoangiogenesis and cell survival. EGF and TGF-α can induce vascular endothelial growth factor mRNA expression (34, 35), thus linking erbB signaling with one mediator of tumor neovessel formation. Moreover, EGFR and ErbB-2/Neu antibodies decrease vascular endothelial growth factor production by receptor-overexpressing tumor cells in vitro and in vivo, thereby reducing the intratumor microvessels (36). However, factor VIII immunostaining revealed a very low content of microvessels in control tumors, indicating that this model is not suited to address the contribution of angiogenesis blockade, as a consequence of interruption of EGFR and ErbB-2/Neu signaling.
Overexpression of ErbB-2/Neu leads to activation of the Ras/MAPK pathway in human breast tumor cell lines and carcinomas (5, 6, 37). In mouse fibroblasts, enhanced Ras/MAPK signaling is associated with enhanced cell cycle progression by mediating the degradation of the Cdk inhibitor p27 and inducing expression of G1 cyclins (ref. 38 and references therein). On the contrary, blockade of EGFR and ErbB-2/Neu kinases in this study eliminated MAPK activity, up-regulated p27, and decreased cyclin D1 tumor levels. This occurred simultaneously with complete inhibition of BrdUrd incorporation and reduction of p-Rb in the tumors. Furthermore, the untreated tumor lysates, which contained constitutively active EGFR and ErbB-2/Neu kinases, degraded recombinant p27 ex vivo, which was prevented by (i) depletion of the proteasome, (ii) treatment of mice with AG-1478 before tumor harvesting, (iii) the MEK1/2 inhibitor U-0126 in vitro, or (iv) a T187A mutation in p27. It has been shown that degradation of p27 by cyclin E/Cdk2 depends on phosphorylation of Thr-187 (32). Kawada et al. (31) reported that MAPK can phosphorylate recombinant p27 in vitro and disrupt its association with Cdk2. The data presented suggest that active MAPK (from transgenic tumors) also can phosphorylate wild-type p27 but not a T187A mutant (Fig. 6c), implying that T187 also may be a target for MAPK-induced phosphorylation and the subsequent degradation of p27.
Our data also suggest a contribution of tumor Cdk2 activity to oncogene-induced cell cycle progression via degradation of p27. The temporally direct correlation of Cdk2 and MAPK activities under conditions of activation or inhibition of EGFR-ErbB-2/Neu signaling (with AG-1478) suggests that some of the effects on Cdk2 are either secondary to or difficult to dissociate from the changes in MAPK function. For example, by increasing cyclin D1 transcription and protein levels, MAPK assists in the assembly of cyclin D1/Cdk4 complexes potentially sequestering p27 (39, 40). This is supported by the observation that activation of the MEK1/MAPK pathway is not sufficient to trigger degradation of p27 unless cyclin D1 and Cdk4 subunits also are co-overexpressed at levels achieved in cells stimulated by serum. These cyclin D1/Cdk4 complexes then can sequester p27, thus reducing its effective inhibitory threshold on Cdk2 and allowing entry into S phase (41). Second, MAPK may directly contribute to proteasome-mediated degradation of p27 by phosphorylating T187, further reducing the level of p27 available for binding to and inhibiting cyclin E/Cdk2. In turn, at a lower stoichiometry, cyclin E/Cdk2 per se can phosphorylate p27 and target it for further proteasome-mediated degradation (32). We should note that the effect of AG-1478 on the steady-state levels and stability of p27 (Figs. 4b and 5b) probably are not due to a nonspecific inhibitory effect on Cdk2 because the quinazoline's IC50 against Cdk2 is >100 μM (42). Furthermore, addition (in vitro) of AG-1478 to Cdk2 immunoprecipitates from A431 cells did not inhibit their ability to phosphorylate glutathione S-transferase-Rb (42). Hence, these results imply that activated EGFR and ErbB-2/Neu, via MAPK signaling, destabilize p27 in a proteasome-dependent manner, thus contributing to dysregulated cell cycle progression. On the other hand, blockade of ErbB kinases in transgenic tumors with AG-1478 reverses these processes by disabling MAPK (Figs. 4b and 6 a and c), leading to a reduction in cyclin D1, decreased sequestration of p27, and stabilization of p27, which then becomes available for binding to and inhibiting cyclin E/Cdk2, leading to growth arrest.
Whether the stabilization of p27 is required for the observed tumor growth arrest will require further studies in p27-null mice bearing the transforming ErbB-2/Neu and TGF-α protooncogenes. Nonetheless, the data presented have several practical implications. (i) Because of the >80% homology in their kinase domains, EGFR “specific” inhibitors also may be effective against other erbB receptors such as ErbB-2/Neu. (ii) These interventions are cytostatic and can prevent or delay tumors in the absence of host tissue toxicity. Because in situ cellular and biochemical endpoints in tumors were evaluated after 5 days of treatment, we cannot rule out an apoptotic effect after more prolonged therapy. (iii) Tumors with loss or low levels of p27 and/or gene amplification at the cyclin D1 locus, phenotypes that occur in human breast cancers (43, 44), may not respond as well to inhibitors of the EGFR and ErbB-2/Neu kinases compared with cancers with normal levels of p27 and cyclin D1. (iv) Finally, MAPK may be an obligatory target that anti-EGFR and anti-ErbB-2/Neu interventions may have to disable to exert an antitumor effect. MAPK activity and expression are increased in some human breast tumors independent of EGFR or ErbB-2/Neu (45). Therefore, tumors harboring hyperactive MAPK secondary to stimulation by kinases other than EGFR or ErbB-2/Neu may be less sensitive to anticancer approaches targeted to ErbB receptors.
Acknowledgments
We thank Teresa Dugger, Galina Bogatcheva, and Sandy Olson for outstanding technical support and Gillian Cropp for the HPLC analysis of plasma samples. The scientific input from Dr. Michele Pagano is enormously appreciated. This work is supported by National Institutes of Health Grants R01 CA80195 (C.L.A.) and R01 CA46413 (R.J.C.), a Clinical Investigator Award from the Department of Veteran Affairs (C.L.A.), the Vanderbilt University Medical Center Cell Imaging Resource (Grant DK20593), and Vanderbilt–Ingram Cancer Center Support Grant CA68485. A.E.G.L. is the recipient of a Susan G. Komen Fellowship Award.
Abbreviations
- MMTV
mouse mammary tumor virus
- TGF-α
transforming growth factor α
- EGFR
epidermal growth factor receptor
- HH1
histone H1
- MAPK
mitogen-activated protein kinase
- Cdk
cyclin-dependent kinase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160564197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160564197
References
- 1.Ullrich A, Coussens L, Hayflick J S, Dull T J, Gray A, Tam A W, Lee J, Yarden Y, Libermann T A, et al. Nature (London) 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 2.Bargmann C I, Hung M C, Weinberg R A. Nature (London) 1986;319:226–230. doi: 10.1038/319226a0. [DOI] [PubMed] [Google Scholar]
- 3.Plowman G D, Colouscou J-M, Whitney G S, Green J M, Carlton G W, Foy L, Neubauer M G, Shoyab M. Proc Natl Acad Sci USA. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plowman G D, Green J M, Culouscou J M, Carlton G W, Rothwell V M, Buckley S. Nature (London) 1993;366:473–475. doi: 10.1038/366473a0. [DOI] [PubMed] [Google Scholar]
- 5.Riese D J, Stern D F. BioEssays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 6.Alroy I, Yarden Y. FEBS Lett. 1997;410:83–86. doi: 10.1016/s0014-5793(97)00412-2. [DOI] [PubMed] [Google Scholar]
- 7.Bargmann C I, Hung M C, Weinberg R A. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 8.Weiner D B, Kokai Y, Wada T, Cohen J A, Williams W V, Greene M I. Oncogene. 1989;4:1175–1183. [PubMed] [Google Scholar]
- 9.Hynes N E, Stern D F. Biochim Biophys Acta. 1996;1198:165–184. doi: 10.1016/0304-419x(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 10.Karunagaran D, Tzahar E, Beerli R R, Chen X, Graus-Porta D, Ratzkin B J, Seger R, Hynes N E, Yarden Y. EMBO J. 1996;15:254–264. [PMC free article] [PubMed] [Google Scholar]
- 11.Graus-Porta D, Beerli R R, Daly J M, Hynes N E. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokai Y, Myers J N, Wada T, Brown V I, LeVea C M, Davis J G, Dobashi K, Greene M I. Cell. 1989;58:287–292. doi: 10.1016/0092-8674(89)90843-x. [DOI] [PubMed] [Google Scholar]
- 13.Cohen B D, Kiener P A, Green J M, Foy L, Fell H P, Zhang K. J Biol Chem. 1996;271:30897–30903. doi: 10.1074/jbc.271.48.30897. [DOI] [PubMed] [Google Scholar]
- 14.Bouchard L, Lamarre L, Tremblay P J, Jolicoeur P. Cell. 1989;57:931–936. doi: 10.1016/0092-8674(89)90331-0. [DOI] [PubMed] [Google Scholar]
- 15.Guy C T, Webster M A, Schaller M, Parsons T J, Cardiff R D, Muller W J. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guy T C, Cardiff R D, Muller W J. J Biol Chem. 1996;271:7673–7678. doi: 10.1074/jbc.271.13.7673. [DOI] [PubMed] [Google Scholar]
- 17.Slamon D J, Clark G M, Wong S G, Levin W J, Ullrich A, McGuire W L. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 18.Slamon D J, Godolphin W, Jones L A, Holt J A, Wong S G, Keith D E, Levin W J, Stuart S G, Udove J, Ullrich A, et al. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 19.Cobleigh M A, Vogel C L, Tripathy D, Robert N J, Scholl S, Fehrenbacher L, Wolter J M, Paton V, Shak S, Lieberman G, et al. J Clin Oncol. 1999;17:2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 20.Muller W J, Arteaga C L, Muthuswamy S K, Siegel P M, Webster M A, Cardiff R D, Meise K S, Li F, Halter S A, Coffey R J. Mol Cell Biol. 1996;16:5726–5736. doi: 10.1128/mcb.16.10.5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arteaga C L, Ramsey T T, Shawver L K, Guyer C A. J Biol Chem. 1997;272:23247–23254. doi: 10.1074/jbc.272.37.23247. [DOI] [PubMed] [Google Scholar]
- 22.Ciardiello F, McGeady M L, Kim N, Basolo F, Hynes N, Langton B C, Yokozaki H, Saeki T, Elliott J W, Masui H, et al. Cell Growth Differ. 1990;1:407–420. [PubMed] [Google Scholar]
- 23.Favata M F, Horiuchi K Y, Manos E J, Daulerio A J, Stradley D A, Feeser W S, Van Dyk D E, Pitts W J, Earl R A, Hobbs F, et al. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 24.Arteaga C L, Koli K M, Dugger T C, Clarke R. J Natl Cancer Inst. 1999;91:46–53. doi: 10.1093/jnci/91.1.46. [DOI] [PubMed] [Google Scholar]
- 25.Dixit M, Yang J L, Poirier M C, Price J O, Andrews P A, Arteaga C L. J Natl Cancer Inst. 1997;89:365–373. doi: 10.1093/jnci/89.5.365. [DOI] [PubMed] [Google Scholar]
- 26.Matsui Y, Halter S A, Holt J T, Hogan B L, Coffey R J. Cell. 1990;61:1147–1155. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 27.Krane I M, Leder P. Oncogene. 1996;12:1781–1788. [PubMed] [Google Scholar]
- 28.Loda M, Cukor B, Tam S W, Lavin P, Fiorentino M, Draetta G F, Jesup J M, Pagano M. Nat Med. 1999;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 29.Lavoie J N, Rivard N, L'Allemain G, Pouyssegur J. Prog Cell Cycle Res. 1996;2:49–58. doi: 10.1007/978-1-4615-5873-6_5. [DOI] [PubMed] [Google Scholar]
- 30.Rivard N, Boucher M J, Asselin C, L'Allemain G. Am J Physiol. 1999;277:C652–C664. doi: 10.1152/ajpcell.1999.277.4.C652. [DOI] [PubMed] [Google Scholar]
- 31.Kawada M, Yamagoe S, Murakami Y, Suzuki K, Mizuno S, Uehara Y. Oncogene. 1997;15:629–637. doi: 10.1038/sj.onc.1201228. [DOI] [PubMed] [Google Scholar]
- 32.Sheaff R, Groudine M, Gordon J, Roberts J, Clurman B. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- 33.Levitzki A, Gazit A. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 34.Goldman C K, Kim J, Wong W L, King V, Brock T, Gillespie G Y. Mol Biol Cell. 1993;4:121–133. doi: 10.1091/mbc.4.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gille J, Swerlick R A, Caughman S W. EMBO J. 1997;16:750–759. doi: 10.1093/emboj/16.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petit A M, Rak J, Hung M C, Rockwell P, Goldstein N, Fendly B, Kerbel R S. Am J Pathol. 1997;151:1523–1530. [PMC free article] [PubMed] [Google Scholar]
- 37.Janes P W, Daly R J, deFazio A, Sutherland R L. Oncogene. 1994;9:3601–3608. [PubMed] [Google Scholar]
- 38.Malumbres M, Pellicer A. Front Biosci. 1998;3:d887–d912. doi: 10.2741/a331. [DOI] [PubMed] [Google Scholar]
- 39.Cheng M, Olivier P, Diehl J A, Fero M, Roussel M F, Roberts J M, Sherr C J. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherr C J, Roberts J M. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 41.Cheng M, Sexl V, Sherr C J, Roussel M F. Proc Natl Acad Sci USA. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Busse D, Doughty R S, Ramsey T T, Russell W E, Price J O, Flanagan W M, Shawver L K, Arteaga C L. J Biol Chem. 2000;275:6987–6995. doi: 10.1074/jbc.275.10.6987. [DOI] [PubMed] [Google Scholar]
- 43.Clurman B E, Porter P. Proc Natl Acad Sci USA. 1998;95:15158–15160. doi: 10.1073/pnas.95.26.15158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall M, Peters G. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 45.Sivaraman V S, Wang H, Nuovo G J, Malbon C C. J Clin Invest. 1997;99:1478–1483. doi: 10.1172/JCI119309. [DOI] [PMC free article] [PubMed] [Google Scholar]