

Abstract
Spinal muscular atrophy (SMA), a common motor neuron disease in humans, results from loss of functional survival motor neuron (SMN1) alleles. A nearly identical copy of the gene, SMN2, fails to provide protection from SMA because of a single translationally silent nucleotide difference in exon 7. This likely disrupts an exonic splicing enhancer and causes exon 7 skipping, leading to abundant production of a shorter isoform, SMN2Δ7. The truncated transcript encodes a less stable protein with reduced self-oligomerization activity that fails to compensate for the loss of SMN1. This report describes the identification of an in vivo regulator of SMN mRNA processing. Htra2-β1, an SR-like splicing factor and ortholog of Drosophila melanogaster transformer-2, promoted the inclusion of SMN exon 7, which would stimulate full-length SMN2 expression. Htra2-β1 specifically functioned through and bound an AG-rich exonic splicing enhancer in SMN exon 7. This effect is not species-specific as expression of Htra2-β1 in human or mouse cells carrying an SMN2 minigene dramatically increased production of full-length SMN2. This demonstrates that SMN2 mRNA processing can be modulated in vivo. Because all SMA patients retain at least one SMN2 copy, these results show that an in vivo modulation of SMN RNA processing could serve as a therapeutic strategy to prevent SMA.
Proximal spinal muscular atrophy (SMA) is a neurodegenerative disorder with progressive paralysis caused by the loss of α-motor neurons in the spinal cord. With an incidence of 1 in 10,000 live births and a carrier frequency of 1 in 50, SMA is the second most common autosomal recessive disorder and the most frequent genetic cause of infantile death (1). SMA patients are subdivided into types I–III according to age of onset and achieved motor abilities (2). All three forms of proximal SMA are caused by mutations within the telomeric copy of the survival motor neuron gene, SMN1 (3). Some 96.4% of 5q-linked SMA patients show homozygous absence of SMN1 caused by deletions or gene conversions, whereas 3.6% display rare subtle mutations (3, 4). Homozygous absence of SMN2 is found in 5% of control individuals; however, loss of SMN2 has no phenotypic effect (3). SMN1 produces exclusively full-length (FL) SMN mRNA. In contrast, SMN2 expresses dramatically reduced FL and abundant levels of transcript lacking exon 7, SMN2Δ7. SMN2 is retained by all patients and a correlation between the SMN2 protein level and the disease state is established (5, 6). This spliced isoform encodes a truncated, less stable protein with reduced self-oligomerization activity (3, 7, 8). We have shown that inclusion of exon 7 in SMN1-derived transcripts and exclusion of this exon in SMN2-derived transcripts is determined by a single nucleotide difference at position +6 in SMN exon 7 (C in SMN1; T in SMN2). This nucleotide difference is nonpolymorphic in the SMN2 gene and likely disrupts an exonic splicing enhancer (ESE) (9, 10).
The removal of introns and joining of exons is performed by the spliceosome, a macromolecular complex (11) that recognizes splice sites, canonical sequences at the exon/intron border, as well as auxiliary splicing elements, such as ESEs (12). An important group of proteins binding to ESEs are serine/arginine-rich splicing factors (SR) and SR-like proteins (13, 14). They are characterized by serine-arginine-rich domain(s) and RNA-recognition motifs that mediate binding to protein and RNA, respectively. SR and SR-like proteins directly interact with small nuclear ribonucleoprotein-associated proteins, which enhances the recognition of a near splice site (12, 15). ESEs function in regulated as well as constitutively expressed exons. Mutations in ESEs are associated with several human diseases (16).
Genetic analyses of SMN exon 7 identified an AG-rich ESE necessary for the production of FL-SMN transcripts and sufficient for high levels of splicing in a heterologous in vitro splicing system (10). One or more regulatory factors likely function through a direct or indirect association with the SMN exon 7 ESE. The identification of a trans-factor(s) that regulates SMN exon 7 inclusion is a critical step in understanding SMN mRNA processing and the molecular mechanisms of SMA.
Methods
Constructs.
Previously described wild-type SMN minigenes, pSMN1, pSMN2, and the mutated minigene pSMN1ex7Cen (replacement of C with T at position exon 7 + 6 in SMN1 background) contain genomic SMN sequences from SMN exon 6–8 (9). Derivative minigenes, SE1, SE2, SE3, and SE2a-c (Fig. 1) contain mutations within the SMN1 exon 7 ESEs (10). The cDNA of Htra2-β1 and Htra2-β3 (17), Htra2-α (18), SF2/ASF (19), SRp30c (20), U2AF65 (21), SAF-B (22), PTB (23), SLM-2 (24), SF1(25), CLK2 and CLK2-KR (26), and YT521B (27) were subcloned into the mammalian expression vector pEGFP-C2 (CLONTECH) to create green fluorescent protein (GFP)-tagged fusion proteins (17). In addition, cDNA of Htra2-β1 was subcloned as a BamHI–XhoI fragment into an hemagglutinin (HA)-tagged pcDNA3 expression vector. In vitro RNA expression constructs containing SMN exon 7 sequences were constructed by generating double-stranded DNA oligonucleotides corresponding to the indicated sequences: SMN1, SMN2, SMNmSE2 (contains mutations in SE2 as indicated in Fig. 1) and SE2 × 3 (contains wild-type SE2 three times). Sequences were cloned into pSP72 between XbaI and HindIII sites, and plasmids then were linearized with XhoI for T7 in vitro transcription reactions to generate sense RNA.
Figure 1.
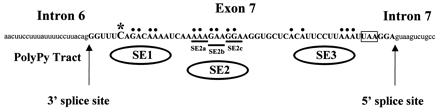
RNA sequence of the SMN exon 7 region indicating exon 7 sequences (bold capital letters) and adjacent intron sequences (lowercase letters). The nucleotide difference between SMN1 and SMN2 in exon 7 at position +6 (*), splice sites, SMN stop codon (boxed), and mutations used in Fig. 3 A–C are indicated. Base pairs indicated with dots and underlining were mutated to “U” residues in the identified regions SE1, SE2, SE3, and SE2a-c (8).
In Vivo Splicing.
In vivo splicing assays were performed as described (28). In brief, 1 μg of the indicated SMN minigene was used together with increasing amounts of pEGFP-Htra2-β1 or the indicated pEGFP-construct. The appropriate amount of empty pEGFP-C2 vector was added to ensure that equal amounts of DNA were transfected. HEK293 cells and NIH 3T3 murine fibroblasts were transfected by using calcium phosphate and Superfect transfection reagent (Qiagen, Hilden, Germany), respectively. RNA was isolated 24 h (HEK293) and 36 h (NIH 3T3) posttransfection by using the E.Z.N.A. total RNA kit (Peqlab, Erlangen, Germany). Reverse transcription was performed in a total volume of 8.5 μl by using 2 μl RNA, 1 μl oligo(dT) (0.5 mg/ml), 1× first-strand buffer (Life Technologies, Karlsruhe, Germany), 100 mM DTT, 10 mM dNTP, 50 units SuperScript II RT (Life Technologies), and 7 units RNase inhibitor (Amersham Pharmacia) at 42°C for 60 min. To ensure that only plasmid-derived SMN transcript was detected, subsequent amplification was performed with a vector-specific forward primer (pCI-forw: 5′-GGT GTC CAC TCC CAG TTC AA) and the SMN-specific reverse primer SMNex8-rev (SMNex8-rev: 5′-GCC TCA CCA CCG TGC TGG). This was necessary because all cell lines contain endogenous SMN. As an internal control the hypoxanthine phosphoribosyltransferase (HPRT) gene was simultaneously amplified by using the primers HPRT-Fw 5′-AAG GAG ATG GGA GGC CAT, and HPRT-Rev 5′-GTT GAG AGA TCA TCT CCA CCA AT. To ensure quantitative measurements during the linear phase, 22 cycles (annealing: 55°C for 30 sec; extension: 72°C for 1 min) were performed (29, 30). PCR products were resolved on 10% PAA gels and visualized by silver staining. PCR amplifications performed with 25 cycles and PCR products resolved on ethidium bromide-stained agarose gels resulted in similar ratios. The ratio of FL-SMN to SMNΔ7 was densitometrically determined by using One-DScan software (MWG Biotec, Ebersberg, Germany). Representative results are shown from multiple separate experiments.
RNA-Protein Binding.
Sense strand RNA corresponding to exon 7 from SMN1, SMN2, SMN1 with mutated SE2, or three wild-type copies of SE2 (SE2 × 3), were transcribed and uniformly labeled with all four [α-32P] NTPs in vitro. RNAs were incubated on ice with 10, 20, or 30 μg of HeLa nuclear extract or 30 μg S100 fraction, which lacks Htra2-β1, with 1× splicing buffer for 15 min before the addition of heparin (final concentration 2 mg/ml) (31). Samples were UV-irradiated (480,000 μJ/cm2), digested with RNaseA (20 μg) and RNaseT1 (40 units), and resolved by 10% SDS/PAGE.
Biotin-conjugated RNA corresponding to SE2 × 3 was reacted with 100 μg of cellular extract from human cervical carcinoma cells C33a (32) transiently transfected with a HA-Htra2-β1 expression construct for 20 min on ice before the addition of streptavidin beads for 30 min on ice. Bound fractions were washed repeatedly in ice-cold PBS with 75 mM NaCl and 0.1% NP-40 and analyzed by Western blotting using 12CA5 α-HA mAb (Santa Cruz Biotechnology). Competitor RNAs were synthesized in vitro and added to HA-Htra2-β1 extracts before the addition of biotin-conjugated SE2 × 3 at 4, 10, and 20 molar excess.
Note, in vivo splicing patterns were similar in numerous cell and tissue types, including C33a (9, 10); therefore this cellular extract was a suitable context in which to examine factor binding to the SE2 element in SMN exon 7.
Results
To identify factors that participate in regulating alternative splicing of SMN2 exon 7, the effects of several mRNA processing factors were tested in transfection assays for their ability to stimulate the inclusion of SMN2 exon 7 and production of higher levels of FL-SMN2 transcripts in vivo. Factors tested included SR domain-containing proteins: Htra2-β1, Htra2-β3 (17), Htra2-α (18), SF2/ASF (19), SRp30c (20) and U2AF65 (21), SAF-B (22) and PTB (23), K homology (KH) domain containing proteins SLM-2 (24), and SF1 (25); the SR protein kinase CLK2 (26) and its inactive form CLK2-KR (26) as well as the splicing associated glutamic acid/arginine-rich protein YT521B (27). In vivo splicing assays were performed in human HEK293 cells, in which increasing amounts (0–3 μg) of expression vectors encoding the individual splicing factors were cotransfected with 1 μg of pSMN2 or pSMN1ex7Cen minigenes. Importantly, splicing of the two minigenes recapitulated the mRNA processing of the endogenous SMN2 gene (9). Of the 13 regulatory factors tested, only Htra2-β1, an SR-like protein that functions through ESEs containing AG-rich motifs (33), concomitantly decreased SMN2Δ7 and increased FL-SMN2 production. Numerous repetitions of this experiment verified a ≈2.7-fold increase of FL-SMN2 transcript at the highest concentration of Htra2-β1 examined (Fig. 2A). These results were in dramatic contrast to normal SMN2 splicing patterns in which <30% of SMN2-derived transcripts were FL.
Figure 2.
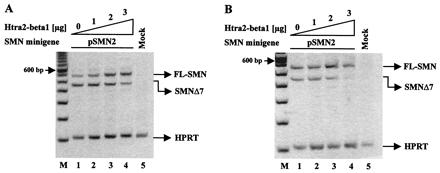
Stimulation of FL-SMN expression by Htra2-β1 in (A) human HEK293 cells and (B) murine NIH 3T3 fibroblasts. (A) Increasing amounts of Htra2-β1 (0–3 μg) were transiently cotransfected with 1 μg of the pSMN2 minigene in HEK293 cells. pSMN2 recapitulated the splicing pattern of endogenous SMN2, with a ratio of 26.5% (27.7 ± 7.4%) FL-SMN2 to 73.5% (72.3 ± 7.4%) SMN2Δ7 (lane 1). Transient expression of Htra2-β1 led to abundant accumulation of FL-SMN2 in a concentration-dependent manner (lanes 2–4); FL-SMN2 increases to 54.4% (57.9 ± 4.7%) (lane 3) and 72.0% (71.0 ± 8.2%) or 2.7 fold increase of FL-SMN2 (lane 4). M = 100-bp ladder (Life Technologies). Similar results were obtained with a synthetic SMN hybrid gene pSMN1ex7Cen, which recapitulates the splicing pattern of SMN2 (data not shown). (B) Increasing amounts of Htra2-β1 (0–3 μg) transiently cotransfected with a constant amount (1 μg) of pSMN2 minigene into murine NIH 3T3 fibroblasts resulted in a ratio of 55.8% (53.3 ± 3.4%) FL-SMN2 to 44.2% (46.7 ± 3.4%) SMN2Δ7 (lane 1). Transient expression of Htra2-β1 led to increased levels of FL-SMN2 in a concentration-dependent manner (lanes 2–4); FL-SMN2 increased to 77.6% (75.8 ± 2.2%) (lane 3) and up to 79.5% (84.8 ± 7.9%) or 1.4-fold increase (lane 4). However, compared with human, high concentration of Htra2-β1 in murine cells seems to have an inhibitory effect on the transcription level from the minigene. The ratio of the SMN transcripts was determined by semiquantitative analyses using the One-DScan software (MWG Biotec). As an internal control part of the HPRT gene was amplified in a multiplex PCR.
We performed similar in vivo splicing assays in murine cells to determine whether the splicing regulation observed in human cells was species-specific. This was important because an SMA transgenic mouse exists in which the human SMN2 gene has replaced the murine Smn gene (34, 35). The identification of factors that stimulate FL-SMN expression could serve as therapeutic agents in an SMA model by altering normal SMN2 mRNA processing to produce predominantly FL-SMN2. Cotransfection of increasing amounts of Htra2-β1 with the SMN2 minigene into murine NIH 3T3 fibroblasts conferred high levels of FL-SMN2 expression and lower levels of SMN2Δ7, comparable to the splicing pattern observed in human HEK293 cells (Fig. 2B). Importantly, the mouse and human Htra2-β1 proteins are identical. In the absence of cotransfected Htra2-β1, the ratio of FL-SMN2 to SMN2Δ7 from the SMN2 minigene differs between murine and human cells (55.8 to 44.2 in murine cells; 26.5 to 73.5 in human cells). However, transient Htra2-β1 expression affects splicing within both cellular contexts similarly by up-regulating FL and down-regulating exon 7 skipping from SMN2. In murine cells a decrease in transcription was observed from the minigene at the highest Htra2-β1 concentration, likely because of squelching (Fig. 2B).
Htra2-β1 binds (GAA)-oligomeric sequences in vitro; however, in vivo targets have not been identified. The SE2 element within SMN exon 7 is an AG-rich ESE essential for FL-SMN expression in vivo (10), and sequence inspection showed that SE2 could represent a putative Htra2-β1 binding motif. To determine whether an intact SMN exon 7 SE2 region was necessary for the stimulatory activity of Htra2-β1, similar in vivo splicing assays were performed with a series of templates containing various mutations in exon 7 on an SMN1 background. Introduction of mutations on SMN1 background rather than an SMN2 background simplified assaying of the effect of the various mutations on the binding of Htra2-β1 to the ESE and the capacity of Htra2-β1 to reverse SMN2Δ7 in FL-SMN2 in vivo. A disruption of the putative Htra2-β1 binding site within SE2 would likely abrogate the stimulatory effect of Htra2-β1. Similarly, disruptions of other elements not essential for SE2 binding and function may be compensated by the overexpression of Htra2-β1. In the absence of cotransfected Htra2-β1, the mutant SE1 and SE3 minigenes resulted in decreased amounts of FL-SMN expression, whereas mutation of SE2 dramatically reduced expression to nearly undetectable levels (Fig. 3A), consistent with previous results (10). In a dose-dependent manner, cotransfection of Htra2-β1 restored higher levels of FL-SMN1 expression to the SE1 and especially to the SE3 mutant minigene (Fig. 3A, lanes 2, 3, 7, and 9). Disruption of the AG-rich element, SE2, could not be overcome to any detectable level even at the highest amount of cotransfected Htra2-β1 (Fig. 3A, lanes 5 and 6). The requirement for an intact SE2 element also was observed in the expression patterns from minigenes that contained double ESE mutations. Cotransfection of Htra2-β1 could not stimulate FL-SMN expression from pSMN1 minigenes that contained the SE2 mutation in conjunction with a mutation in either SE1 or SE3 (Fig. 3B, lanes 6, 7, 9, and 10). This suggests that SE2 contains the critical sequence motif through which Htra2-β1 functions and likely binds SMN exon 7, an observation consistent with Htra2-β1 recognition elements identified by in vitro SELEX (systematic evolution of ligands by exponential enrichment) (33). Because mutations within SE1 partially affect, whereas mutations in SE3 do not affect the ability of Htra2-β1 to up-regulate FL-SMN1 and down-regulate SMN1Δ7 transcripts, suggest that SE1 could contribute to the Htra2-β1:SE2 enhancer complex.
Figure 3.
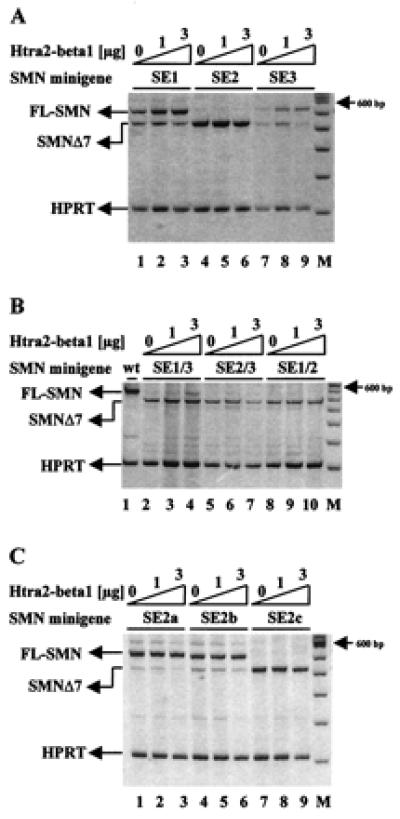
Identification of the Htra2-β1-responsive element within SMN exon 7. (A) SMN1 minigenes containing mutations within SE1 or SE3 showed an increased level of SMN1Δ7 in the absence of Htra2-β1 (lanes 1 and 7). Cotransfection of Htra2-β1 revealed an 1.7-fold increase of FL expression to the SE1 mutant template (lanes 3) and a 2.9-fold increase FL expression to the SE3 mutant (lane 9). Disruption of the AG-rich element, SE2, resulted only in SMN1Δ7 expression (lane 4) and could not be overcome even at the highest concentration of Htra2-β1 (lane 6). (B) Combined disruption of SE1 and SE3 resulted in exclusive SMN1Δ7 expression in the absence of cotransfected Htra2-β1 (lane 2) and could be only slightly compensated by cotransfection of 3 μg Htra2-β1 (lane 4). The SE2 mutation with either SE3 (lanes 5–7) or SE1 (lanes 8–10) mutations resulted exclusively in SMN1Δ7, indicating that SE2 is necessary for the inclusion of exon 7 and the site through which Htra2-β1 functions. (C) Smaller disruptions dividing SE2 into three subdomains (SE2a, SE2b, and SE2c) resulted in low levels of SMN1Δ7 from minigenes with SE2a (lane 1) and SE2b (lane 4) mutations. Transient expression of 3 μg Htra2-β1 resulted in a partly reversion of SMN1Δ7 in FL-SMN1 shown by a 50% decrease of SMN1Δ7 transcript (lanes 3 and 6). The disrupted SE2c minigene resulted in the exclusive expression of SMN1Δ7 (lane 7) and also could not be compensated by transient expression of Htra2-β1 (lanes 8 and 9).
To more finely map the Htra2-β1 responsive element(s), a series of minigenes containing smaller sequential substitution mutations within the AG-rich SE2 region were used (10). These mutations replaced three consecutive bases from the AG-rich region with “UUU” residues, dividing SE2 into three subdomains: SE2a, SE2b, and SE2c (Fig. 1) (10). Disruption of SE2a and SE2b had a modest effect on FL-SMN expression, whereas mutation of SE2c resulted in high levels of SMN1Δ7 and undetectable levels of FL-SMN, consistent with previous results (10). Cotransfection of Htra2-β1 reversed about half of SMNΔ7 into FL-SMN1 in SE2a and SE2b, but had no affect to SE2c (Fig. 3C). These results demonstrate that the mutation in SE2c had the most dramatic affect on FL-SMN expression and may represent the primary Htra2-β1 binding motif.
To identify proteins associated with the SE2 enhancer, uniformly α32P-NTP-labeled RNA and splicing competent HeLa nuclear extract were mixed and UV cross-linked. In these experiments we used templates of SMN exon 7 RNAs corresponding to SMN1, SMN2, and the mutant SMN1 exon 7 containing a disrupted SE2 element (SMNmSE2), as well as three wild-type copies of SE2 (SE2 × 3). After digestions of these complexes with RNaseA and RNaseT1, proteins cross-linked to RNA were resolved by SDS/PAGE. A complex of ≈40 kDa was detected with SMN1 and SMN2 but not with the SMNmSE2 template (Fig. 4A). Because SMN exon 7 contains multiple regulatory elements that bind to trans-factors, a template that was comprised exclusively of three copies of the SE2 element also was examined. Importantly, SE2 × 3 RNA also resulted in a similarly sized ≈40-kDa complex. The mobility of this SE2-dependent complex was consistent with the mobility of endogenous Htra2-β1 (36). To confirm that Htra2-β1 bound to SE2, cellular extracts containing transiently transfected HA-tagged Htra2-β1 were mixed with biotin-conjugated RNA corresponding to SE2 × 3. Reactions were bound to streptavidin beads, washed, and analyzed by Western blotting using 12CA5, an anti-HA mAb. Transiently transfected HA-tagged Htra2-β1 bound SE2 × 3 RNA and was specifically blocked by in vitro-transcribed SE2 × 3 RNA but not RNA corresponding to SMN exon 7 with the SE2 mutation (Fig. 4B). Taken together, these results demonstrated that Htra2-β1 promotes exon 7 usage by binding to the SE2 enhancer to stimulate FL-SMN expression.
Figure 4.
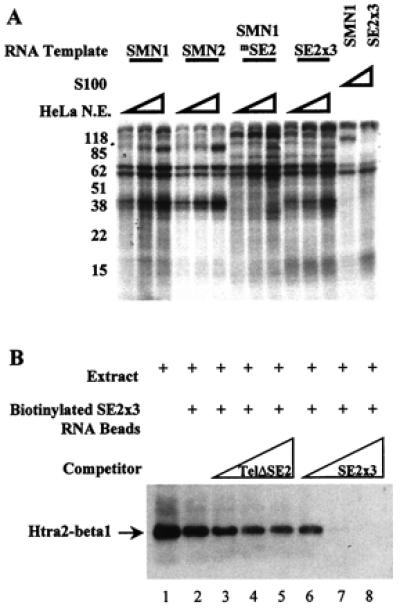
Htra2-β1 binds the SMN exon 7 ESE SE2. (A) Cross-linking of cellular factors to SMN exon 7 RNA. Sense strand RNA corresponding to exon 7 from SMN1, SMN2, SMN1 with the SE2 mutation (SMN1mSE2), or three wild-type copies of SE2 (SE2 × 3), were transcribed and uniformly labeled with all four α-32P NTPs in vitro. Increasing amounts (10, 20, or 30 μg) of splicing competent HeLa nuclear extract or 30 μg of the S100 fraction were reacted with the RNAs under splicing conditions for 15 min before heparin was added (final concentration 2 mg/ml). Samples were UV-irradiated, digested with RNaseA (20 μg) and RNaseT1 (40 units), and resolved by 10% SDS/PAGE. (B) In vitro binding of Htra2-β1 and SMN-derived RNA. C33a cellular extract containing transiently transfected HA-tagged Htra2-β1 (lane 1) binds efficiently to RNA corresponding to three copies of the AG-rich SE2 element (lane 2) when detected by HA-specific antibodies. The RNA-protein interaction of Htra2-β1 and SE2 × 3 RNA was poorly blocked by the addition of increasing amounts of SMN exon 7 RNA that contained the SE2 mutation (lanes 3–5). SE2 × 3:Htra2-β1 binding was efficiently competed by the addition of increasing amounts of RNA corresponding to three wild-type copies of SE2 (lanes 6–8).
Discussion
Recently, it has been established that the critical difference between the SMA-determining gene, SMN1, and the nearly identical copy gene, SMN2, is the ability of SMN1 to produce high levels of FL-SMN. Genetic studies identified a single nucleotide difference between the two genes that was critical for FL expression as well as an AG-rich ESE (SE2) within SMN exon 7 (9, 10). This study was further supported by the complete sequence analysis of both SMN genes, that showed that the C-to-T transition in exon 7 is the only significant change (37). This report identified Htra2-β1 as a factor that modulated SMN2 expression in vivo, reverting the splicing pattern of SMN2 comparable to the normal SMN1 gene and produced high levels of FL-SMN. Furthermore, this work underscores the importance of the SE2 element because Htra2-β1 was shown to require the presence of an intact SE2 element and that Htra2-β1 bound specifically to the AG-rich SE2 element. The stimulatory effect of exon 7 inclusion was specific and not a consequence of a global stimulation of cellular RNA processing because Htra2-β1 has been previously shown to exert no effect or even repress expression of heterologous exons (38). However, Htra2-β1 likely regulates multiple in vivo targets not identified here, because it is expressed in various tissues including brain (36).
SMN is the first example of a human gene whose splicing is regulated by Htra2-β1. Previously it has been shown by in vitro SELEX procedures that Htra2-β and Htra2-α bind oligo(GAA)-containing sequences (33). Nevertheless, in vivo experiments with the SMN2 minigene demonstrated a high degree of specificity; only Htra2-β1 stimulated the up-regulation of FL-SMN2, whereas Htra2-β3 isoform and Htra2-α failed to stimulate FL expression (data not shown).
Htra1-β is the Drosophila melanogaster Tra2 ortholog (17, 18), the latter being involved in a critical sex-differentiation pathway that is regulated by alternative splicing (39). Consistent with the classification of Drosophila Tra2 as a nonessential SR-like protein, homozygous loss of this gene transforms chromosomally female flies into sterile, but otherwise developmentally normal, males (40). Like its Drosophila ortholog, mammalian Tra2 isoforms may be nonessential as they are absent from some tissues (17) and cell types (36).
Because Htra2-β1 efficiently stimulates high levels of FL-SMN transcripts from SMN2, these results have implications for potential SMA therapies. In this context it would be interesting to know whether endogenous FL-SMN2 transcripts also are up-regulated by Htra2-β1. Therefore, stable transfected cell lines, preferably from an SMA patient, need to be established to ensure simultaneous transcription of SMN2 and Htra2-β1 temporally and spatially.
Typically, only SMN1 produces sufficient levels of SMN to provide protection from disease; however, all SMA patients retain at least a single copy of SMN2. In this report a splicing factor “converted” the mRNA processing pattern of SMN2 to nearly that of SMN1. Based on these data, transgenic SMA mice carrying human SMN2 (34, 35) could be used to determine whether modulating factors such as Htra2-β1 stimulate FL-SMN expression and influence SMA development. The construction of transgenic mice with the genotype Smn−/−;SMN;Htra2-β1 under a strong neuron-specific promotor may address the question of whether SMA can be cured by overexpression of Htra2-β1 as well as whether overexpressed Htra2-β1 may affect other transcripts.
Using cellular extracts or overexpression systems it was demonstrated that splice site choice is influenced by the concentration of SR proteins (14, 41, 42). SR proteins are enriched in nuclear structures called speckles. It is assumed that reversible phosphorylation of SR proteins from speckles controls their release into the nucleoplasma, and the resulting changes in local SR protein concentration regulate alternative splicing (43, 44). Tra2-β was proposed to be part of a splicing regulatory complex conserved from Drosophila to human (18, 36). Under physiological conditions, its isoform ratios (36) and intracellular concentrations (45) are regulated by neuronal activity and nerve injury, respectively, indicating its role in splice site selection in vivo. Therefore, drugs aimed at releasing specifically Htra2-β1 from its nuclear storage sites could result in the production of FL-SMN2 and might be of therapeutic use.
Various hormones, in particular sex steroid hormones, have been shown to regulate alternative splicing (46). Given the fact that rare cases of SMA, particularly in females, show homozygous loss of SMN1, but no SMA phenotype (47, 48), one could speculate that a sex-specific factor might differentially regulate Htra2-β1 in these females and prevents them from getting SMA. Taken together, our results open the exciting possibility that SMA may be treated by agents that promote high levels of FL expression from SMN2.
Acknowledgments
We are very grateful to Families of SMA for promoting research and discussion on SMA. Funding of these studies was provided by the Deutsche Forschungsgemeinschaft and Families of SMA (to B.W.), the Muscular Dystrophy Association and Families of SMA (to E.J.A.), and the Max-Planck-Society and Deutsche Forschungsgemeinschaft (to S.S.). C.L.L. was supported by a New Investigator Development Award from the Muscular Dystrophy Association.
Abbreviations
- SMA
spinal muscular atrophy
- SMN
survival motor neuron gene
- ESE
exonic splicing enhancer
- FL
full-length
- SR
serine/arginine-rich splicing factor
- GFP
green fluorescent protein
- HA
hemagglutinin
- HPRT
hypoxanthine phosphoribosyltransferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160181697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160181697
References
- 1.Pearn J. Lancet. 1980;i:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 2.Munsat T L, Davies K E. Neuromusc Disord. 1992;2:423–428. doi: 10.1016/s0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 3.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 4.Wirth B. Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 6.Coovert D D, Le T T, McAndrew P E, Strasswimmer J, Crawford T O, Mendell J R, Coulson S E, Androphy E J, Prior T W, Burghes A H. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 7.Lorson C L, Strasswimmer J, Yao J M, Baleja J D, Hahnen E, Wirth B, Le T, Burghes A H, Androphy E J. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 8.Pelizzoni L, Charroux B, Dreyfuss G. Proc Natl Acad Sci USA. 1999;96:11167–11172. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorson C L, Hahnen E, Androphy E J, Wirth B. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorson C L, Androphy E J. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 11.Krämer A. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 12.Tacke R, Manley J L. Curr Opin Cell Biol. 1999;11:358–362. doi: 10.1016/S0955-0674(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 13.Fu X-D. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 14.Manley J L, Tacke R. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- 15.Wu J Y, Maniatis T. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 16.Cooper T A, Mattox W. Am J Hum Genet. 1997;61:259–266. doi: 10.1086/514856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayler O, Cap C, Stamm S. Genomics. 1998;53:191–202. doi: 10.1006/geno.1998.5471. [DOI] [PubMed] [Google Scholar]
- 18.Dauwalder B, Amaya-Manzanares F, Mattox W. Proc Natl Acad Sci USA. 1996;93:9004–9009. doi: 10.1073/pnas.93.17.9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayeda A, Krainer A R. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 20.Screaton G R, Caceres J F, Mayeda A, Bell M V, Plebanski M, Jackson D G, Bell J I, Krainer A R. EMBO J. 1995;14:4336–4349. doi: 10.1002/j.1460-2075.1995.tb00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamore P D, Patton J G, Green M R. Nature (London) 1992;355:609–614. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 22.Nayler O, Straetling W, Bourquin J-P, Stagljar I, Lindemann L, Jasper H, Hartmann A M, Fackelmeyer F O, Ullrich A, Stamm S. Nucleic Acids Res. 1998;26:3542–3549. doi: 10.1093/nar/26.15.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gil A, Sharp P A, Jamison S F, Garcia-Blanco M A. Genes Dev. 1991;5:1224–1236. doi: 10.1101/gad.5.7.1224. [DOI] [PubMed] [Google Scholar]
- 24.Di Fruscio M, Chen T, Richard S. Proc Natl Acad Sci USA. 1999;96:2710–2715. doi: 10.1073/pnas.96.6.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Bruderer S, Rafi Z, Xue J, Milburn P J, Kramer A, Robinson P J. EMBO J. 1999;18:4549–4559. doi: 10.1093/emboj/18.16.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nayler O, Stamm S, Ullrich A. Biochem J. 1997;326:693–700. doi: 10.1042/bj3260693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann A M, Nayler O, Schwaiger F W, Obermeier A, Stamm S. Mol Biol Cell. 1999;10:3909–3926. doi: 10.1091/mbc.10.11.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoss O, Stoilov P, Hartmann A M, Nayler O, Stamm S. Brain Res Protein. 1999;4:383–394. doi: 10.1016/s1385-299x(99)00043-4. [DOI] [PubMed] [Google Scholar]
- 29.Gavrilov D K, Shi X, Das K, Gilliam T C, Wang C H. Nat Genet. 1998;20:230–231. doi: 10.1038/3030. [DOI] [PubMed] [Google Scholar]
- 30.Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schöneborn S, Wienker T, Zerres H. Am J Hum Genet. 1999;64:1340–1356. doi: 10.1086/302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Z M, He P J, Baker C. J Virol. 1997;71:9096–9107. doi: 10.1128/jvi.71.12.9096-9107.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strasswimmer J, Lorson C L, Breiding D E, Chen J J, Le T, Burghes A H, Androphy E J. Hum Mol Genet. 1999;8:1219–1926. doi: 10.1093/hmg/8.7.1219. [DOI] [PubMed] [Google Scholar]
- 33.Tacke R, Tohyama M, Ogawa S, Manley J L. Cell. 1998;93:139–148. doi: 10.1016/s0092-8674(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh-Li H, Chang J, Jong H, Li H. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 35.Monani U R, Sendtner M, Coovert D D, Parsons D W, Andreassi C, Le T T, Jablonka S, Schrank B, Rossol W, Prior T W, et al. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 36.Daoud R, Berzaghi M, Siedler F, Hübener M, Stamm S. Eur J Neurosci. 1999;11:788–802. doi: 10.1046/j.1460-9568.1999.00486.x. [DOI] [PubMed] [Google Scholar]
- 37.Monani U R, Lorson C L, Parsons D W, Prior T W, Androphy E J, Burghes A H M, McPherson J D. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 38.Stamm S, Casper D, Hanson V, Helfman D M. Mol Brain Res. 1999;64:108–118. doi: 10.1016/s0169-328x(98)00313-1. [DOI] [PubMed] [Google Scholar]
- 39.Baker B S. Nature (London) 1989;340:521–524. doi: 10.1038/340521a0. [DOI] [PubMed] [Google Scholar]
- 40.Baker B S, Ridge K A. Genetics. 1980;94:383–423. doi: 10.1093/genetics/94.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cáceres J, Stamm S, Helfman D M, Krainer A R. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- 42.Hanamura A, Caceres J F, Mayeda A, Franza B R, Jr, Krainer A R. RNA. 1998;4:430–444. [PMC free article] [PubMed] [Google Scholar]
- 43.Misteli T, Cáceres J F, Spector D L. Nature (London) 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 44.Nayler O, Schnorrer F, Stamm S, Ullrich A. J Biol Chem. 1998;273:34341–34348. doi: 10.1074/jbc.273.51.34341. [DOI] [PubMed] [Google Scholar]
- 45.Kiryu-Seo S, Matsuo N, Wanaka A, Ogawa S, Tohyama M, Kiyama H. Mol Brain Res. 1998;62:220–223. doi: 10.1016/s0169-328x(98)00255-1. [DOI] [PubMed] [Google Scholar]
- 46.Guivarc'h D, Vincent J D, Vernier P. Endocrinology. 1998;139:4213–4221. doi: 10.1210/endo.139.10.6246. [DOI] [PubMed] [Google Scholar]
- 47.Hahnen E, Forkert R, Marke C, Rudnik-Schoneborn S, Schonling J, Zerres K, Wirth B. Hum Mol Genet. 1995;4:1927–1933. doi: 10.1093/hmg/4.10.1927. [DOI] [PubMed] [Google Scholar]
- 48.Cobben J M, van der Steege G, Grootscholten P, de Visser M, Scheffer H, Buys C H. Am J Hum Genet. 1995;57:805–808. [PMC free article] [PubMed] [Google Scholar]