

Abstract
Smad4/DPC4 (deleted in pancreatic carcinoma, locus 4) is a tumor suppressor gene lost at high frequency in cancers of the pancreas and other gastrointestinal organs. Smad4 encodes a key intracellular messenger in the transforming growth factor β (TGF-β) signaling cascade. TGF-β is a potent inhibitor of the growth of epithelial cells; thus, it has been assumed that loss of Smad4 during tumor progression relieves this inhibition. Herein, we show that restoration of Smad4 to human pancreatic carcinoma cells suppressed tumor formation in vivo, yet it did not restore sensitivity to TGF-β. Rather, Smad4 restoration influenced angiogenesis, decreasing expression of vascular endothelial growth factor and increasing expression of thrombospondin-1. In contrast to the parental cell line and to control transfectants that produced rapidly growing tumors in vivo, Smad4 revertants induced small nonprogressive tumors with reduced vascular density. These data define the control of an angiogenic switch as an alternative, previously unknown mechanism of tumor suppression for Smad4 and identify the angiogenic mediators vascular endothelial growth factor and thrombospondin-1 as key target genes.
The Smad4/DPC4 (deleted in pancreatic carcinoma, locus 4) gene is a candidate tumor suppressor gene (1) that is frequently inactivated in pancreatic (1–3), biliary (4, 5), and colorectal (6–8) tumors. Germ-line mutations of Smad4 have been reported in familial juvenile polyposis (9, 10). Smad4 belongs to the evolutionarily conserved family of Smad proteins that are crucial intracellular mediators of signals from the transforming growth factor β (TGF-β) superfamily of cytokines (11–13), multifunctional proteins that govern intercellular interactions in development and homeostasis and regulate proliferation, differentiation, and cell death (14–17). All members of this family signal through serine threonine kinase receptor complexes that propagate signals to a variety of receptor-regulated Smad proteins. Activated receptor-regulated Smads then form heteromeric complexes with the common partner Smad4/DPC4 (co-Smad), and these heteromeric complexes translocate to the nucleus, where they function as transcriptional regulators (11–13, 18). As the single mammalian co-Smad identified so far, Smad4 thus plays a pivotal role affecting all TGF-β superfamily signal pathways. Smad4's tumor suppressor function is usually studied in the context of TGF-β signaling, as the roles of other family members, the bone morphogenetic proteins and activins, in human tumorigenesis are just beginning to be unraveled.
TGF-β can play complex roles in tumorigenesis behaving as a potent tumor suppressor at early stages of carcinogenesis as well as a tumor promoter at late stages (for review, see refs. 19 and 20). Inhibition of epithelial cell growth is the most prominent of TGF-β's biological activities, which also include induction of differentiation or apoptosis and maintenance of genomic stability. When tumor cells lose sensitivity to TGF-β growth inhibition, the excess TGF-β that results may act on tumor cells and stromal cells to facilitate invasion and metastasis, induce angiogenesis, and suppress antitumor immune responses.
Although the multifunctional nature of the TGF-β superfamily suggests several potential mechanisms by which loss of Smad4 may contribute to tumorigenesis, previous analyses of putative tumor suppressor functions of Smad4 have concentrated on its presumed role as a mediator of TGF-β's tumor suppressive functions. Reexpression of Smad4 in human cancer cell lines growing in vitro has shown the expected results, such as restoration of TGF-β signaling, induction of apoptosis, and/or growth inhibition (21–25), but these effects in vitro have not been reported to be associated with suppression of tumorigenicity in vivo.
Herein, we report that restoration of Smad4 to human pancreatic carcinoma cells blocked their ability to grow progressively as tumors in vivo. Unexpectedly, Smad4 reexpression did not restore sensitivity to growth inhibition by TGF-β. Rather, Smad4 decreased the expression of vascular endothelial growth factor (VEGF) and increased levels of the angiogenesis inhibitor thrombospondin-1 (TSP-1) causing the cells to switch from potently angiogenic to antiangiogenic in vitro and in vivo. These findings suggest that, in pancreatic adenocarcinoma cells, Smad4 exerts at least a portion of its tumor suppressive function by controlling an angiogenic switch and identify VEGF and TSP-1 as tumor-relevant targets of the ubiquitous Smad4 signaling cascade.
Materials and Methods
Cell Culture and Gene Transfer.
Hs766T human pancreatic adenocarcinoma cells were obtained from the American Type Culture Collection. Cells were maintained in DMEM supplemented with antibiotics and 10% (vol/vol) FCS (GIBCO). The full-length coding sequence of Smad4/DPC4 was cloned into the pBK-CMV expression vector (Stratagene) as described (26). Cells were transfected by a standard calcium phosphate coprecipitation method. Stable transfectants were selected with geneticin (0.2 mg/ml; GIBCO) and isolated by ring cloning.
RNA Isolation, Northern Hybridization, and RNase Protection Assay.
RNA was isolated from parental cells and transfectants by acid phenol extraction. Northern blots and hybridization were performed as described (27). Signal intensities were quantitated by PhosphorImager analysis (Packard). RNase protection assays were performed with RiboQuant MultiTemplate Sets (PharMingen) according to the manufacturer's instructions.
Western Blot Analysis.
Cell lysis, Western blots, and detection of DPC4/Smad4 were performed as described (26). For VEGF Western blots, cell culture supernatants were concentrated with Centricon3 tubes, and 10 μg of total protein per lane was separated under nonreducing conditions. VEGF was detected with primary mouse monoclonal antibody no. 26503.11 (R & D Systems). For TSP-1 Western blots, supernatants were concentrated with Centricon100 tubes, and 1 μg per lane was electrophoresed under reducing conditions. TSP-1 was detected with antibody A6.1 (Oncogene Research Products).
In Vitro and in Vivo Growth Assay.
For in vitro growth assays 1 × 105 cells were plated per 60-mm dish. Cells were counted from duplicate plates every 2 or 3 days as indicated. For analysis of TGF-β-mediated growth inhibition, 2 × 105 cells per 60-mm dish were shifted to low serum medium [2% (vol/vol) FCS] plus TGF-β (5 ng/ml; R & D Systems) 1 day after plating and were re-fed with fresh medium plus TGF-β every 3 days. Cell numbers were determined from triplicate plates as indicated. For tumorigenicity assays, suspensions of 5 × 106 cells in a volume of 0.1 ml of PBS were injected s.c. into the flanks of 7- to 8-week-old female athymic nude mice (BALB/c01aHsd-nu/nu). Tumor diameter was measured in two dimensions with a caliper twice a week.
Angiogenesis Assays.
Endothelial cell migration assays were performed in a modified Boyden chamber as described (28). Briefly, endothelial cells were plated in the lower wells of the chamber and allowed to attach to the undersurface of a gelatinized membrane (5-μm pore size). Serum-free conditioned medium (20 μg/ml) in DMEM with 0.1% BSA and other test substances or neutralizing antibodies were added to the upper wells of the chamber and incubated for 4 h at 37°C in 8% CO2. The filters were stained, and the number of cells migrating to the top of the membrane per 10 high-powered fields was counted. Results are reported as percentage of induced migration compared with standard inducer (VEGF at 0.1 ng/ml) after subtraction of background migration toward vehicle (0.1% BSA). All samples were tested in quadruplicate.
In vivo neovascularization was assayed in the rat cornea as described (28). Briefly, Hydron pellets containing conditioned medium (200 μg/ml) alone or with inducer (1 ng/ml VEGF or 100 ng/ml basic fibroblast growth factor) or neutralizing antibody to VEGF (5 μg/ml) or TSP-1 (antibody A4.1 at 20 μg/ml) were implanted into the avascular cornea of anesthetized female rats (Harlan–Sprague–Dawley, Indianapolis), approximately 1.0–1.5 mm from the limbus. On day 7, the animals were killed and perfused with colloidal carbon; the eyes were fixed in 10% (vol/vol) buffered formalin; and the corneas were excised and photographed. Vigorous in-growth of vessels toward the pellet was scored as a positive response. This protocol was approved by the Animal Care and Use Committee of Northwestern University.
Histology and Antibody Staining.
Histological sections were prepared from fresh-frozen nude mouse tumors, fixed, and subjected to immunohistochemistry with rat anti-mouse CD31 antibody (PharMingen). Detection of primary antibody was performed with mouse anti-rat secondary antibody and alkaline phosphatase antialkaline phosphatase staining. Tissue sections (two to three per tumor) from controls (24 analyzed) and revertant tumors (34 analyzed) were stained, and endothelial cords (width < 5 μm; length > 20 μm), capillaries (diameter < 10 μm), medium-sized vessels (diameter 10–50 μm), and large vessels (diameter > 50 μm) were counted in three to five arbitrarily chosen optical fields per section at the tumor periphery.
Results
Stable Reexpression of Smad4/DPC4 in Hs766T Pancreatic Carcinoma Cells.
To examine mechanisms of Smad4-mediated tumor suppression, we performed reconstitution experiments of Smad4 in human pancreatic adenocarcinoma cells. Stable transfections of Hs766T, cells null for Smad4 (1), with a Smad4 expression construct and an empty vector control yielded similar numbers of G418 resistant clones, indicating lack of growth inhibitory or toxic effects of the transferred Smad4 under normal culture conditions. Northern blot analysis revealed reexpression of Smad4 in about two-thirds of the clones derived from expression vector transfections. Expression levels of Smad4 were comparable among the positive clones (Fig. 1A), and Western blot analysis confirmed stable reexpression of the Smad4 protein, albeit at levels that were lower than endogenous protein levels in Paca44 cells, which served as a representative positive control (Fig. 1B). Smad4 reexpression did not exert pronounced effects on the morphology of Hs766T cells and was not sufficient to trigger significant responses in transient transfections of p3TPlux or SBE6lux reporters. Transient overexpression of Smad4 and co-overexpression of Smad3 plus Smad4 in Hs766T cells, however, increased luciferase expression from the SBE6lux reporter by 2-fold and 7-fold in the absence of TGF-β and by 4-fold and 15-fold on TGF-β induction, respectively (data not shown).
Figure 1.
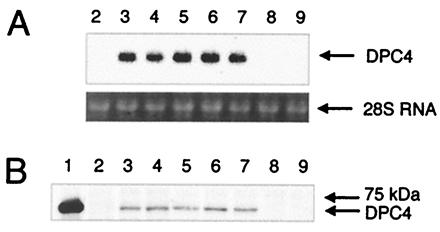
Reexpression of the human Smad4/DPC4 in the human pancreatic adenocarcinoma cell line Hs766T. (A) Northern blot analysis of total RNA from the parental cell line and stable transfectants hybridized with a human Smad4/DPC4 cDNA probe. Lane 2, Hs766T parental cell line; lanes 3–7, Smad4/DPC4 reconstituted clones D2, D4, D5, D8, and D9; lanes 8 and 9, negative control clones K3 and K6. (B) Western blot analysis for the human Smad4/DPC4 protein on total protein extracts. Lane 1, human pancreatic adenocarcinoma cell line Paca44 as control for endogenous Smad4/DPC4; lane 2, Hs766T parental cell line; lanes 3–7, Smad4/DPC4 reconstituted clones; lanes 8 and 9, negative control clones.
Smad4/DPC4-Reexpressing Cells Retain TGF-β Resistance.
Because Hs766T cells, like many tumor cells, express high levels of endogenous TGF-β (ref. 29 and unpublished data), one might expect retarded cell growth of Smad4-positive cells through restoration of an autocrine loop. Smad4-expressing and nonexpressing clones, however, possessed virtually identical population doubling times in culture (Fig. 2A), consistent with the fact that transfection efficiencies were not reduced. Moreover, addition of high concentrations of recombinant active TGF-β1 to growth media also failed to inhibit the growth of either Smad4-expressing or nonexpressing clones in vitro (Fig. 2B), whereas growth of TGF-β responsive HaCat control cells was reduced by 66%. In line with this result, TGF-β in Hs766T cell clones failed to induce cyclin-dependent kinase inhibitor p15, increased levels of cyclin-dependent kinase inhibitor p21 by less than 2-fold, only, irrespective of their Smad4 status, and did not affect the ratio of hypophosphorylated pRB, in contrast to responsive HaCat cells (data not shown). Expression analysis of further components of the signal transduction pathway mediating TGF-β-induced growth inhibition revealed significant expression levels of Smad2, Smad3, and TGF-βRII in Hs766T cells, comparable to those in a panel of TGF-β responsive and nonresponsive cell lines (data not shown). TGF-βRI specific signals, however, were barely detectable in Smad4- positive or -negative transfectants and in the parental cell line (Fig. 2C), consistent with a previous report (29).
Figure 2.
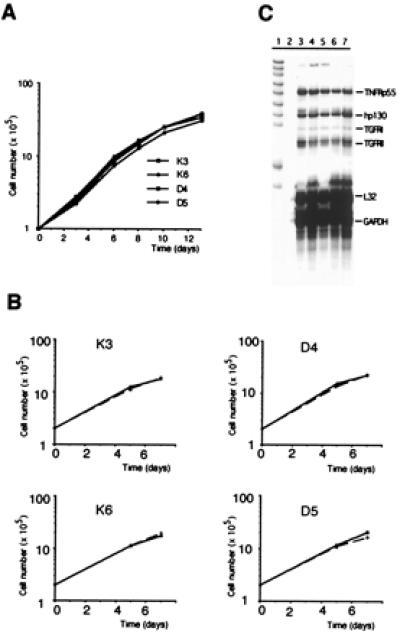
Analysis of growth and TGF-β response in vitro and expression analysis of TGF-β receptors. (A) Growth of Smad4/DPC4-reexpressing cell clones and negative controls cultured in serum-supplemented growth medium. (B) Growth of Smad4/DPC4-reexpressing cells and negative control cells incubated in the absence (continuous bar) or presence (hatched bar) of TGF-β1 (5 ng/ml) under reduced serum concentrations. (A and B) Cells were plated in duplicate (A) or triplicate (B) for each time point, and results were confirmed in at least two independent experiments. Note that error bars for standard deviations are shown in B but are too narrow to be resolved. (C) Expression of TGF-βRI and TGF-βRII as analyzed by RNase protection assay. Lane 1, probe; lane 2, tRNA; lane 3, Hs766T parental cell line; lanes 4 and 5, Smad4/DPC4 negative control clones K3 and K6; lanes 6 and 7, Smad4/DPC4-reconstituted clones D5 and D8.
Reexpression of Smad4/DPC4 Induces Suppression of Tumorigenicity.
Tumorigenic potential in vivo is governed by complex processes and has many requirements in addition to deregulated growth control (for review see ref. 30). To assess directly Smad4's tumor-suppressive function, we performed tumorigenicity assays in nude mice. Smad4-negative control cells when injected s.c. yielded rapidly growing tumors, and animals had to be killed in less than 3 weeks. In contrast, tumors derived from Smad4-expressing clones showed a strong decrease of median tumor mass at the same time (Fig. 3A), whereas tumor-take rates were not reduced. Moreover, these small tumors came to a halt after reaching 3–5 mm in diameter. Median tumor size did not change significantly in the following 5 weeks when the experiment was terminated (Fig. 3B). These results show that, although Smad4 was not sufficient to restore TGF-β sensitivity to Hs766T cells in vitro, it was adequate to inhibit and to halt tumor growth.
Figure 3.
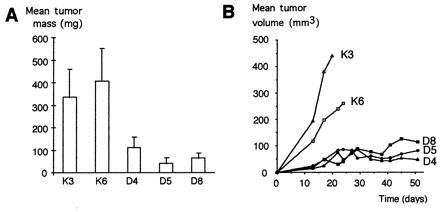
Suppression of tumor growth in vivo. (A) Mean mass of tumors derived from control clones K3 and K6 and Smad4/DPC4-reexpressing clones D4, D5, and D8 (mean of eight tumors each). All mice were killed, and tumors were prepared and weighed by 17 days after s.c. injection, when tumors from the control clones reached 10 mm in diameter. (B) Time course of tumor growth derived from control clones and Smad4/DPC4-reexpressing clones in an independent experiment (tumor volume = length × width2/2, mean of six tumors from each clone).
Smad4/DPC4 Controls Expression Levels of VEGF and TSP-1.
This arrest of tumor growth at a small size suggested that angiogenesis might be limiting. Expansion of solid tumors critically depends on an adequate vascular supply, and the switch to an angiogenic phenotype is a prerequisite for tumor growth and metastasis (31–33). Therefore, we investigated expression levels of prominent angiogenesis regulators in Smad4-expressing and nonexpressing cell clones. Strikingly, steady-state mRNA levels of the potent angiogenesis activator VEGF were constitutively reduced in all of the Smad4 reexpressing clones by 2- to 3-fold (Fig. 4A). In addition, mRNA levels of the potent angiogenesis inhibitor TSP-1 were increased approximately 3-fold in Smad4-reexpressing clones. These results were confirmed in cells cultivated in serum-free medium. VEGF and TSP-1 protein levels in cell culture supernatants were shifted correspondingly (Fig. 4B), suggesting that Smad4 may regulate expression of VEGF and TSP-1 in a reciprocal manner.
Figure 4.
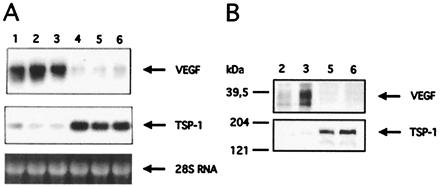
Smad4/DPC4-mediated shifts in VEGF and TSP-1 expression levels. (A) Northern blot with total RNA was hybridized with a human VEGF cDNA probe, stripped, and rehybridized with a TSP-1-specific cDNA probe. Lane 1, Hs766T parental cell line; lanes 2 and 3, Smad4/DPC4-negative clones K3 and K6; lanes 4–6, Smad4/DPC4-reconstituted clones D4, D5, and D8. The difference in expression levels was confirmed with at least three independent RNA preparations each. Quantification of RNA levels repeatedly revealed a reduction of the VEGF steady-state mRNA level by a factor of two to three and induction of TSP-1 expression by a factor of approximately three in Smad4/DPC4-reexpressing clones grown in full medium or incubated in serum-free medium. (B) VEGF and TSP-1 Western blots with protein from conditioned media. Lanes 2 and 3, Smad4/DPC4-negative clones K3 and K6; lanes 5 and 6, Smad4/DPC4-reconstituted clones D5 and D8. The signals correspond to dimeric VEGF165 and trimeric TSP-1.
Smad4/DPC4 Controls an Angiogenic Switch.
To test the possibility that Smad4 controls angiogenic activity when expressed in revertant cells, the angiogenic potential of Smad4 reconstituted and control cells was assessed directly by testing conditioned media in endothelial cell migration assays in vitro and in rat cornea neovascularization assays in vivo (Figs. 5 and 6). Supernatants of Smad4-negative cells strongly stimulated endothelial cell migration (Fig. 5A) and induced corneal neovascularization (Fig. 6). Addition of a VEGF-neutralizing antibody reduced these activities to background levels (Figs. 5B and 6), confirming VEGF as an essential angiogenesis inducer in Smad4-negative cells. In contrast, conditioned media of Smad4 expressing cells failed to induce endothelial cell migration or neovascularization. They were also capable of inhibiting angiogenesis induced by VEGF or basic fibroblast growth factor. However, when TSP-1 was depleted by the addition of a neutralizing antibody, moderate angiogenic activities reappeared, which could be further elevated by the addition of VEGF (Figs. 5C and 6). Thus, Smad4-mediated suppression of angiogenic activity was exerted through two mechanisms: reduction of the major angiogenesis inducer VEGF and induction of the angiogenesis inhibitor TSP-1.
Figure 5.
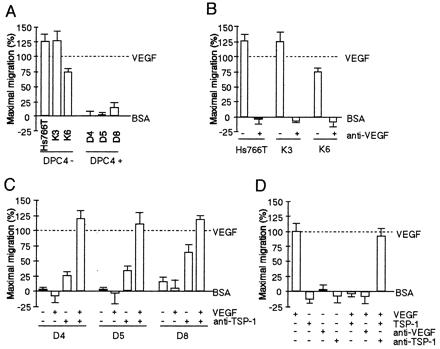
Activation of endothelial cell migration by Smad4/DPC4-negative control cells is due to VEGF, and inhibitory activity of Smad4/DPC4-reconstituted cells is due to TSP-1. (A) Conditioned media (20 μg/ml) were tested to evaluate induction of migration in Smad4/DPC4-negative and Smad4/DPC4-positive cells. (B) Conditioned media from Smad4/DPC4-negative cells were tested in the absence or presence of neutralizing anti-VEGF antibody (at 5 μg/ml). (C) Conditioned media from Smad4/DPC4-positive cells were tested in the absence or presence of neutralizing anti-TSP-1 antibody (20 μg/ml) with or without addition of recombinant VEGF (0.1 ng/ml). (D) Controls demonstrating the effect of neutralizing antibodies and recombinant proteins. All samples were tested in quadruplicate. Bars, standard errors converted to percentages. All experiments were repeated and gave virtually identical results.
Figure 6.
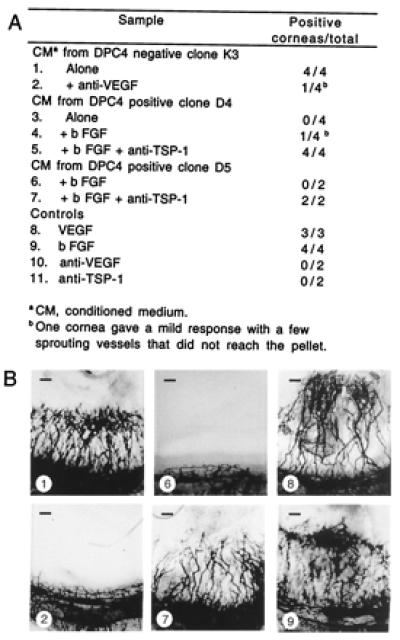
In vivo angiogenic activity of Smad4/DPC4-reconstituted clones and negative control. (A) Hydron pellets containing conditioned medium (200 μg/ml) alone or with inducer (1 ng/ml VEGF or 100 ng/ml basic fibroblast growth factor) or neutralizing antibody to VEGF (5 μg/ml) or TSP-1 (antibody A4.1 at 20 μg/ml) were implanted into the avascular rat cornea. Vigorous in-growth of vessels from the limbus toward the pellet by 7 days was scored as a positive response. (B) Representative photos of corneal responses from A. (Bars = 200 μm.)
To determine whether the antiangiogenic phenotype of Smad4-revertant cells was retained in vivo, we analyzed vascularization of nude mouse tumors by immunostaining. Importantly, whereas endothelial cords were slightly more frequent in Smad4-positive tumors, numbers of capillaries were reduced slightly, and densities of medium-sized and large vessels were reduced significantly to 68% and 50% of controls, respectively (Fig. 7). Thus, Smad4 effects on VEGF and TSP-1 expression may contribute to reduced tumor growth through diminished vascular supply.
Figure 7.
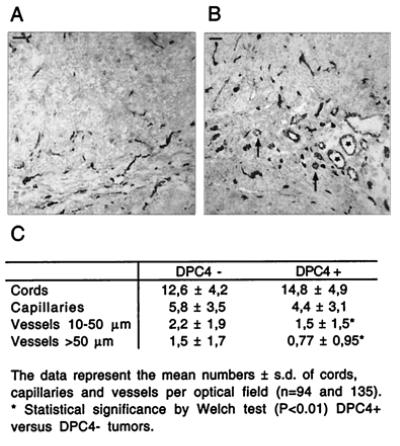
Vascularization of nude mouse tumors. CD31-immunostained section through a tumor derived from Smad4/DPC4-negative control clone K3 (A) and derived from Smad4/DPC4-positive clone D5 (B). Asterisks in A depict large vessels (diameter > 50 μm), and medium-sized vessels (diameter 10–50 μm) are indicated by arrows. (Bars = 100 μm.) (C) Quantification of cords, capillaries, and vessels in Smad4-negative and Smad4-positive Hs766T nude mouse tumors.
Discussion
Despite extensive knowledge of the biochemical properties of Smad proteins, little is known about how the loss of Smad4 function contributes to the tumorigenic process. Herein, we report the surprising result that Smad4-mediated suppression of tumorigenicity in human pancreatic adenocarcinoma cells is not due to restoration of TGF-β antiproliferative responses. Instead, reexpression of Smad4 induced a shift in angiogenic phenotype from inducing to inhibiting, defining an alternative mechanism of Smad4-mediated tumor suppression.
Neovascularization through the induction of angiogenesis is a prerequisite for growth of solid tumors beyond a microscopic size (31, 32). In normal adult tissues, the physiological control of angiogenesis is regulated by a balance of inducers and inhibitors (32), and expression of these regulators is tightly controlled. There is accumulating evidence that tumor suppressors may act in part through suppression of angiogenesis. p53 (34–37), p16 (38), and the von Hippel–Lindau gene (39, 40) have all been implicated in VEGF down-regulation, and wild-type p53 increases expression of TSP-1 (31, 34, 37, 41). Herein, we show that reexpression of Smad4 in pancreas carcinoma cells at the same time decreased expression levels of VEGF and increased the expression of TSP-1. Both changes were essential and, together, were sufficient to shift the angiogenic phenotype of Hs766T cells to antiangiogenic. The angiogenic switch seen in vitro was maintained in vivo, where Smad4-expressing cells formed small tumor nodules with reduced vascularization.
Both VEGF and TSP-1 are produced by the normal pancreas. VEGF overexpression is a frequent finding in pancreatic carcinomas and has been associated with the dismal prognosis of these tumors and their propensity for hematogenous metastasis in the early phase of the disease (42). TSP-1 is a potent inhibitor of neovascularization when soluble (43). It is also deposited in the matrix, where it may function as a shield that hinders blood vessel penetration into a tumor (44). Although TSP-1 is produced in pancreatic islets, where it activates TGF-β (45), its expression in pancreatic adenocarcinomas has not yet been investigated.
Stable transfer of Smad4 was not sufficient to restore a TGF-β growth inhibitory response in Hs766T cells. There could be several reasons for this outcome. Hs766T cells harbor a variety of molecular alterations like mutations of the tumor suppressor genes p53 and p16 (46, 47) that would be retained in Smad4 revertants and may underlie retained resistance. Moreover, low expression levels of TGF-βRI as well as low expression levels of Smad4 as achieved in the reconstituted Hs766T cells may limit antiproliferative responses. Oncogenic activation of Ki-ras in concert with Smad4 inactivation may cause TGF-β resistance in SW480 colon carcinoma cells (48), but this reason seems unlikely here, because Hs766T cells retain wild-type copies of Ki-ras.
Although our results do not disprove a role of Smad4 as mediator of TGF-β growth inhibitory responses, a number of recent findings further support our contention that loss of Smad4 may enhance tumorigenicity by mechanisms that are independent of growth inhibition by TGF-β. Smad4 is dispensable for a growth-inhibitory response in murine fibroblasts (49) and in human pancreas carcinoma cells (50). Smad4 loss is often late in tumor progression, after normal growth constraints would have been overcome. The frequency of Smad4 inactivation in human colon tumors is strongly increased on acquisition of metastasis (8) and loss of Smad4 expression occurs late in the development of pancreatic adenocarcinomas, coincident with the onset of carcinomatous change (51). Experimental evidence from Smad4-reconstituted SW480 colon carcinoma cells (26) and from APC/DPC4 compound knockout mice (52) also indicates that Smad4 may be related to the acquisition of invasive and metastatic potential.
It is not yet clear how Smad4 controls VEGF and TSP-1. The VEGF promoter does harbor several potential Smad-binding sequences, an SBE-related sequence (53) at position −1,225, two overlapping CAGA boxes (54) in the 5′ untranslated region, and a GC-rich sequence closely related to a FAST2/DPC4-binding goosecoid promoter element (55) in the proximal promoter region; thus, it could be controlled directly. The TSP-1 promoter is devoid of currently known Smad-binding elements. Because of the versatile ways by which Smad proteins can regulate gene expression (56), indirect mechanisms are conceivable.
It has been suggested previously, that Smad4, expressed at limiting levels, may function to sense and interpret multiple TGF-β related signals (57). In addition, as multiple signaling pathways impinge on Smad family members, Smad4 may fine tune the cellular response in a multistimulatory environment (56). In line with this hypothesis, preliminary experimental evidence indicates that stable restoration of Smad4 in Hs766T cells may alter intricate signal integration mechanisms. On treatment with recombinant TGF-β1, expression levels of VEGF and TSP-1 were increased in both Smad4-positive and Smad4-negative Hs766T clones; the Smad4-mediated differences in expression levels, however, were retained. In conclusion, the future understanding of Smad4 signaling may require models that are based on a signaling network rather than a single linear pathway. Detailed analysis of the expression profile and activation status of cytokines, receptors, Smad proteins, and interacting transcription factors in tumor cells are needed to approach that goal.
Our results do not support the prevailing view that Smad4's tumor suppressor function in pancreatic carcinoma cells primarily resides in its capability to mediate TGF-β growth inhibitory responses. Rather, they suggest that the acquisition of TGF-β resistance and loss of Smad4 may be independent consecutive events in the tumorigenic process, each contributing to successful tumor development—one freeing tumor cells of direct growth constraints by TGF-β, the other freeing the tumor of indirect growth constraints imposed through limited vascularization. Pancreatic adenocarcinomas characteristically express increased levels of TGF-β isoforms, associated with alterations of the extracellular matrix, induction of fibrosis and angiogenesis, and suppression of the immune response (58). It will be interesting in the future to investigate Smad4 involvement in mediating and/or counteracting these effects and to decipher the underlying molecular mechanisms.
Acknowledgments
We thank C. Lechleiter and T. Haberland for animal care, S. Scory for statistical analysis, N. Müller for image analysis, J. Folkman for bovine capillary endothelial cells, S. Blass-Kampmann for help with tumorigenicity assays, P. Lorenz for help with antibody production, and M. F. Rajewsky for support. This work was supported by grants from the Dr. Mildred Scheel Stiftung für Krebsforschung (10-1137-HaI), the Deutsche Forschungsgemeinschaft (SCHW 588/2-1), the European Community (Biomed 2 Project, BMH4-CT98-3085) and from the Ruhr-Universität Bochum (FORUM-Programm).
Abbreviations
- DPC4
deleted in pancreatic carcinoma, locus 4
- TGF-β
transforming growth factor β
- VEGF
vascular endothelial growth factor
- TSP-1
thrombospondin-1
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hahn S A, Schutte M, Hoque A T M, Moskaluk C A, da Costa L T, Rozenblum E, Weinstein C L, Fischer A, Yeo C J, Hruban R H, et al. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 2.Schutte M, Hruban R H, Hedrick L, Cho K R, Nadasdy G M, Weinstein C L, Bova G S, Isaacs W B, Cairns P, Nawroz H, et al. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- 3.Bartsch D, Hahn S A, Danichevski K D, Ramaswamy A, Bastian D, Galehdari H, Barth P, Schmiegel W, Simon B, Rothmund S. Oncogene. 1999;18:2367–2371. doi: 10.1038/sj.onc.1202585. [DOI] [PubMed] [Google Scholar]
- 4.Hahn S A, Bartsch D, Schroers A, Galehdari H, Becker M, Ramaswamy A, Schwarte-Waldhoff I, Maschek H, Schmiegel W. Cancer Res. 1998;58:1124–1126. [PubMed] [Google Scholar]
- 5.Goggins M, Shekher M, Turnacioglu K, Yeo C J, Hruban R H, Kern S E. Cancer Res. 1998;58:5329–5332. [PubMed] [Google Scholar]
- 6.Takagi Y, Kohmura H, Futamura M, Kida H, Tanemura H, Shimokawa K, Saji S. Gastroenterology. 1996;111:1369–1372. doi: 10.1053/gast.1996.v111.pm8898652. [DOI] [PubMed] [Google Scholar]
- 7.Thiagalingam S, Lengauer C, Leach F S, Schutte M, Hahn S A, Overhauser J, Willson J K V, Markowitz S, Hamilton S R, Kern S E, et al. Nat Genet. 1996;13:342–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 8.Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, et al. Oncogene. 1999;18:3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 9.Howe J R, Roth S, Ringold J C, Summers R W, Järvinen H J, Sistonen P, Tomlinson I P M, Houlston R S, Bevan S, Mitros F A, et al. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 10.Friedl W, Kruse R, Uhlhaas S, Stolte M, Schartmann B, Keller K M, Jungck M, Stern M, Loff S, Back W, et al. Genes Chromosomes Cancer. 1999;25:403–406. [PubMed] [Google Scholar]
- 11.Massagué J, Hata A, Liu F. Trends Cell Biol. 1997;7:187–192. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- 12.Derynck R, Feng X-H. Biochim Biophys Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- 13.Heldin C-H, Miyazono K, ten Dijke P. Nature (London) 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 14.Roberts A B, Sporn M B. In: Peptide Growth Factors and Their Receptors. Sporn M B, Roberts A B, editors. Heidelberg: Springer; 1990. pp. 419–472. [Google Scholar]
- 15.Sporn M B, Roberts A B. J Cell Biol. 1992;119:1017–1021. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kingsley D M. Genes Dev. 1994;8:133–146. doi: 10.1101/gad.8.2.133. [DOI] [PubMed] [Google Scholar]
- 17.Massagué J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 18.Attisano L, Wrana J. Curr Opin Cell Biol. 1998;10:188–194. doi: 10.1016/s0955-0674(98)80141-5. [DOI] [PubMed] [Google Scholar]
- 19.Akhurst R J, Balmain A. J Pathol. 1999;187:82–90. doi: 10.1002/(SICI)1096-9896(199901)187:1<82::AID-PATH248>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Reiss M. Microbes Infect. 1999;1:1327–1347. doi: 10.1016/s1286-4579(99)00251-8. [DOI] [PubMed] [Google Scholar]
- 21.de Caestecker M P, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts A B, Lechleider R J. J Biol Chem. 1997;272:13690–13696. doi: 10.1074/jbc.272.21.13690. [DOI] [PubMed] [Google Scholar]
- 22.de Winter J P, Roelen B A J, ten Dijke P, van der Burg B, van den Eijnden-van Raaij A J M. Oncogene. 1997;14:1891–1899. doi: 10.1038/sj.onc.1201017. [DOI] [PubMed] [Google Scholar]
- 23.Grau A M, Zhang L, Wang W, Ruan S, Evans D B, Abbruzzese J L, Zhang W, Chiao P J. Cancer Res. 1997;57:3929–3934. [PubMed] [Google Scholar]
- 24.Atfi A, Buisine M, Mazars A, Gespach C. J Biol Chem. 1997;272:24731–24734. doi: 10.1074/jbc.272.40.24731. [DOI] [PubMed] [Google Scholar]
- 25.Dai J L, Bansal R K, Kern S E. Proc Natl Acad Sci USA. 1999;96:1427–1432. doi: 10.1073/pnas.96.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarte-Waldhoff I, Klein S, Blass-Kampmann S, Hintelmann A, Eilert C, Dreschers S, Kalthoff H, Hahn S A, Schmiegel W. Oncogene. 1999;18:3152–3158. doi: 10.1038/sj.onc.1202641. [DOI] [PubMed] [Google Scholar]
- 27.Schwarte-Waldhoff I, Martin W, Willecke K, Schäfer R. Oncogene. 1994;9:899–909. [PubMed] [Google Scholar]
- 28.Polverini P J, Bouck N P, Rastinejad F. Methods Enzymol. 1991;198:440–450. doi: 10.1016/0076-6879(91)98044-7. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin R L, Friess H, Yokoyama M, Lopez M E, Kobrin M S, Büchler M W, Korc M. Int J Cancer. 1996;67:283–288. doi: 10.1002/(SICI)1097-0215(19960717)67:2<283::AID-IJC21>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg R A. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 31.Bouck N, Stellmach V, Hsu S. Adv Cancer Res. 1996;69:135–174. doi: 10.1016/s0065-230x(08)60862-3. [DOI] [PubMed] [Google Scholar]
- 32.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 33.Woodhouse E C, Chuaqui R F, Liotta L A. Cancer. 1997;80:1529–1537. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1529::aid-cncr2>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 34.Dameron K M, Volpert O V, Tainsky M A, Bouck N. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 35.Kieser A, Weich H A, Brandner G, Marme D, Kolch W. Oncogene. 1994;9:963–969. [PubMed] [Google Scholar]
- 36.Mukhopadhyay D, Tsiokas L, Sukhatme V P. Cancer Res. 1995;55:6161–6165. [PubMed] [Google Scholar]
- 37.Volpert O V, Dameron K M, Bouck N. Oncogene. 1997;14:1495–1502. doi: 10.1038/sj.onc.1200977. [DOI] [PubMed] [Google Scholar]
- 38.Harada H, Nakagawa K, Iwata S, Saito M, Kumon Y, Sakaki S, Sato K, Hamada K. Cancer Res. 1999;59:3783–3789. [PubMed] [Google Scholar]
- 39.Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Baron G, Marme D. Cancer Res. 1996;56:2299–2301. [PubMed] [Google Scholar]
- 40.Mukhopadhyay D, Knebelmann B, Cohen H T, Ananth S, Sukhatme V P. Mol Cell Biol. 1997;17:5629–5639. doi: 10.1128/mcb.17.9.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grossfeld G D, Ginsberg D A, Stein J P. J Natl Cancer Inst. 1997;89:219–227. doi: 10.1093/jnci/89.3.219. [DOI] [PubMed] [Google Scholar]
- 42.Itakura J, Ishiwata T, Friess H, Fujii H, Matsumoto Y, Büchler M W, Korc M. Clin Cancer Res. 1997;3:1309–1316. [PubMed] [Google Scholar]
- 43.Dawson D W, Bouck N. In: Antiangiogenic Agents in Cancer. Teicher B A, editor. Totowa, NJ: Humana; 1999. pp. 185–204. [Google Scholar]
- 44.Bleuel K, Popp S, Fusenig N F, Stanbridge E J, Boukamp P. Proc Natl Acad Sci USA. 1999;96:2065–2070. doi: 10.1073/pnas.96.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crawford S E, Stellmach V, Murphy-Ullrich J E, Ribeiro S M, Lawler J, Hynes R O, Boivin G P, Bouck N. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 46.Slebos R J C, Resnick M A, Taylor J A. Cancer Res. 1998;58:5333–5336. [PubMed] [Google Scholar]
- 47.Caldas C, Hahn S A, da Costa L T, Redston M S, Schutte M, Seymour A B, Weinstein C L, Hruban R H, Yeo C J, Kern S E. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 48.Calonge M J, Massagué J. J Biol Chem. 1999;274:33637–33643. doi: 10.1074/jbc.274.47.33637. [DOI] [PubMed] [Google Scholar]
- 49.Sirard C, Kim S, Mirtsos C, Tadich P, Hoodless P A, Itié A, Maxson R, Wrana J L, Mak T W. J Biol Chem. 2000;275:2063–2070. doi: 10.1074/jbc.275.3.2063. [DOI] [PubMed] [Google Scholar]
- 50.Dai J L, Schutte M, Bansal R K, Wilentz R E, Sugar A Y, Kern S E. Mol Carcinog. 1999;26:37–43. doi: 10.1002/(sici)1098-2744(199909)26:1<37::aid-mc5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 51.Wilentz R E, Iacobuzio-Donahue C A, Argani P, McCarthy D M, Parsons J L, Yeo C J, Kern S E, Hruban R H. Cancer Res. 2000;60:2002–2006. [PubMed] [Google Scholar]
- 52.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin M F, Taketo M M. Cell. 1998;92:645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 53.Zawel L, Dai J, Buckhaults S, Zhou S, Kinzler K, Vogelstein B, Kern S. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 54.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier J. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Labbé E, Silvestri C, Hoodless P A, Wrana J L, Attisano L. Mol Cell. 1998;2:109–120. doi: 10.1016/s1097-2765(00)80119-7. [DOI] [PubMed] [Google Scholar]
- 56.Verschueren K, Huylebroeck D. Cytokine Growth Factor Rev. 1999;10:187–199. doi: 10.1016/s1359-6101(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 57.Candia A F, Watabe T, Hawley S H B, Onichtchouk D, Zhang Y, Derynck R, Niehrs C, Cho K W Y. Development (Cambridge, UK) 1997;124:4467–4480. doi: 10.1242/dev.124.22.4467. [DOI] [PubMed] [Google Scholar]
- 58.Korc M. Surg Oncol Clin N Am. 1998;7:25–41. [PubMed] [Google Scholar]