

Abstract
An evaluation of 18 DNA restriction endonucleases for use in terminal-restriction fragment length polymorphism (T-RFLP) analysis was performed by using richness and density indices in conjunction with computer simulations for 4,603 bacterial small-subunit rRNA gene sequences. T-RFLP analysis has become a commonly used method for screening environmental samples for precursory identification and community comparison studies due to its precision and high-throughput capability. The accuracy of T-RFLP analysis for describing a community has not yet been thoroughly evaluated. In this study, we attempted to classify restriction endonucleases based upon the ability to resolve unique terminal-restriction fragments (T-RFs) or operational taxonomic units (OTUs) from a database of gene sequences. Furthermore, we assessed the predictive accuracy of T-RFLP at fixed values of community richness (n = 1, 5, 10, 50, and 100). Classification of restriction endonuclease fidelity was performed by measuring richness and density for the entire database of T-RFs. Further analysis of T-RFLP accuracy for determining richness was performed by iterative, random sampling from the derived database of T-RFs. It became apparent that two constraints were influential for measuring the fidelity of a given restriction endonuclease: (i) the ability to resolve unique sequence variants and (ii) the number of unique T-RFs that fell within a measurable size range. The latter constraint was found to be more significant for estimating restriction endonuclease fidelity. Of the 18 restriction endonucleases examined, BstUI, DdeI, Sau96I, and MspI had the highest frequency of resolving single populations in model communities. All restriction endonucleases used in this study detected ≤70% of the OTUs at richness values greater than 50 OTUs per modeled community. Based on the results of our in silico experiments, the most efficacious uses of T-RFLP for microbial diversity studies are those that address situations where there is low to intermediate species richness (e.g., colonization, early successional stages, biofilm formation).
Microbial ecology studies have come to rely heavily on molecular analyses due to the difficulty arising from exclusively characterizing a natural community by cultivation and microscopy (2, 14, 20, 43). The molecular tool commonly used for examining microbial communities is the small-subunit rRNA gene (SSU rDNA). The SSU rDNA is valuable because it is present in all known extant organisms, it has a relatively low rate of evolution with both conserved and variable regions, and it has been characterized for a broad array of organisms (39, 51). After PCR-based amplification of the gene, the goal is to determine SSU rDNA sequence information and to infer characteristics (e.g., phylogeny and perhaps metabolic capacity) based on descriptions of closely related taxa (16, 18, 29). Characterization of a microbial community by this laborious, sequence-based methodology is often precluded or replaced by a screening technique that elucidates the complexity of a community. There are numerous procedures available to characterize a community of gene amplicons. Terminal-restriction fragment length polymorphism (T-RFLP) analysis is one of the procedures (3, 5, 23) that can be used to track spatial and temporal changes in SSU rDNAs from microbial communities (10, 46; see references 21 and 42 for reviews).
The T-RFLP technique has become a common diagnostic and screening method due to its high sensitivity and ability to rapidly acquire precise data compared to more laborious or imprecise forms of analysis, such as denaturing gradient gel electrophoresis (12, 18, 35, 40) and restriction fragment length polymorphism-amplified ribosomal DNA restriction analysis (1, 7, 17, 37, 44, 48, 50). In the T-RFLP technique one or more fluorescently labeled primers are used during PCR; however, compared to SSU rDNAs, the 5′ or forward fragments are generally more heterogeneous (36). After enzymatic digestion of PCR amplicons, each unique terminal-restriction fragment (T-RF) can be defined as an operational taxonomic unit (OTU) and may often be inferred to be a single population within a community (36). The validity of this inference has not been fully explored yet. Previous models have described restriction endonuclease use in conjunction with the detection of resulting polymorphisms for characterization of diversity both in general (41) and specifically for SSU rDNAs (38). However, neither of these studies dealt with the application to T-RFLP. When T-RFLP is considered, the selection and number of restriction endonucleases should be screened for maximum fidelity of the T-RFs. We define fidelity as the ability of a restriction endonuclease to identify the actual number of SSU rDNA sequence variants derived from a community through an analysis of T-RF size distributions. By choosing the appropriate number and types of restriction endonucleases, an investigator increases the probability that the resulting arrays of T-RF size distributions more accurately reflect the natural diversity of microbial populations within a sampled community.
This study was designed to explore (i) whether sequence variants were more clearly resolved by using selected restriction endonucleases and (ii) to measure the success of restriction endonucleases at detecting sequence variants from model communities that varied in richness. We used traditional diversity measurements and computer simulations to explore the resolving power of select tetrameric restriction endonucleases given a specific set of PCR primers used to amplify an ∼1,460-bp region of the SSU rDNA from organisms belonging to the domain Bacteria. The derived database of T-RFs for each restriction endonuclease was considered a model community, and calculation of richness and density indices provided a measure of the resolving power of a given restriction endonuclease. The fidelity of both restriction endonucleases and the T-RFLP assay under different SSU rDNA richness conditions was measured by performing computer simulation experiments.
MATERIALS AND METHODS
Algorithm to determine PCR amplicon and T-RF length.
A database of available unaligned SSU rDNA sequences (n = 14,870) was acquired from the Ribosomal Database Project (RDP), release 8.1 (6, 30), on 30 July 2001. A computer program was written in Qbasic programming language to perform both in silico PCR screening and restriction endonuclease digestion. The degenerate PCR primers used for this computer analysis were located at Escherichia coli positions 49 to 68F (5′-TNA NAC ATG CAA GTC GNN CG-3′) and 1492 to 1510R (3′-TTC AGC ATT GTT CCA TNG N-5′) (11, 37). In order to acquire a large T-RFLP-compatible SSU rDNA database, sequences exhibiting ≥80% similarity to the primers were selected for further analysis. Exterior ends were trimmed, and PCR amplicon lengths were checked to verify that the DNA sequence sizes were ∼1,460 bp. The selection of restriction endonucleases used (Table 1) was an attempt to screen all readily available tetrameric (4-base cutter) restriction endonucleases. T-RF lengths were calculated by counting the number of string characters from the terminal end to the first cutting site for a given restriction endonuclease. The compiled T-RF lengths were then stored in a new database.
TABLE 1.
Summary of the numbers of OTUs and density indices associated with restriction endonuclease digests from 4,603 SSU rDNA sequences
| Restriction endonuclease | Recognition site | Total no. of OTUs | No. of OTUs in the range from 50 to 500 bp | No. of T-RFs in the range from 50 to 500 bp | D1a,b | D2a,c |
|---|---|---|---|---|---|---|
| Hpy188III | TC^NNGA | 571 (1)d | 320 (2) | 3,129 (11) | 38.5 (1) | 6.29 (1) |
| HhaIe | GCG^C | 563 (2) | 271 (5) | 2,817 (15) | 32.8 (5) | 5.36 (5) |
| DpnIIe | ^GATC | 542 (3) | 255 (9) | 3,430 (9) | 29.8 (10) | 4.87 (10) |
| ScrFI | CC^NGG | 532 (4) | 263 (7) | 2,697 (16) | 31.6 (6) | 5.17 (6) |
| BfaIe | C^TAG | 524 (5) | 244 (12) | 2,876 (14) | 29.3 (11) | 4.79 (11) |
| RsaI | GT^AC | 513 (6) | 253 (10) | 3,011 (12) | 31.0 (7) | 5.06 (7) |
| Tsp509I | ^AATT | 512 (7) | 295 (3) | 2,682 (17) | 36.7 (2b) | 5.99 (2b) |
| MseI | T^TAA | 502 (8) | 235 (14) | 2,247 (18) | 30.3 (9) | 4.96 (9) |
| HpyCH4IVe | A^CGT | 456 (9) | 230 (15) | 3,973 (6) | 24.1 (18) | 3.94 (18) |
| HinfI | G^ANTC | 422 (10) | 248 (11) | 4,103 (3) | 28.4 (12) | 4.64 (12) |
| MspIe | C^CGG | 406 (11) | 322 (1) | 4,027 (5) | 36.7 (2a) | 5.99 (2a) |
| HaeIII | GG^CC | 346 (12) | 241 (13) | 3,956 (7) | 27.6 (14) | 4.51 (14) |
| Hpy1881 | TCN^GA | 345 (13) | 283 (4) | 3,258 (10) | 33.9 (4) | 5.55 (4) |
| BslI | CCN5^NNGG | 315 (14) | 265 (6) | 3,708 (8) | 30.8 (8) | 5.04 (8) |
| AluI | AG^CT | 294 (15) | 210 (18) | 2,993 (13) | 25.5 (15) | 4.18 (15) |
| BstUIe | CG^CG | 292 (16) | 262 (8) | 4,353 (1) | 27.9 (13) | 4.56 (13) |
| DdeI | C^TNAG | 280 (17) | 228 (16) | 4,130 (2) | 24.6 (17) | 4.03 (17) |
| Sau96I | G^GNCC | 273 (18) | 221 (17) | 4,088 (4) | 25.4 (16) | 4.16 (16) |
Calculated for T-RFs for the range from 50 to 500 bp. Data were randomly removed to obtain 2,247 T-RFs per restriction endonuclease in order to remove the effects of sample size.
For a community of 2,247 individuals the range of possible values was 0 to 291.
For a community of 2,247 individuals the range of possible values was 0 to 47.
The numbers in parentheses are ranks.
The restriction endonuclease has one or more known isoschizomers.
Calculation of OTU richness and density.
The database of T-RFs for each restriction endonuclease was considered a model community that was used to evaluate individual restriction endonucleases. Apparent richness was defined as the number of T-RFs with unique sizes resulting in the categorization of OTUs for a given restriction endonuclease. Density indices, which are traditionally misidentified as richness indices, were calculated for each restriction endonuclease-defined community within a desired size range (50 to 500 bp) in order to provide an initial evaluation of the resolving power. D1 is the Margalef index (32) and is calculated as follows: D1 = (S − 1)/ln(n), where S is the number of OTUs and n is the total number of T-RFs. The minimum value of Margalef's index is zero (when the number of OTUs is 1), and the maximum value is (n − 1)/[ln(n)] (when each OTU is represented by one T-RF). D2 is the Menhinick index (34) and is calculated as follows: D2 = S/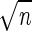 . Menhinick's index approaches zero when there is a high number of individuals but few OTUs and, like Margalef's index, approaches a maximum value when the number of OTUs is equal to the number of individuals. D1 and D2 are sensitive to variations in sample size (26); therefore, data were normalized by randomly removing T-RFs from each restriction endonuclease profile until all sample sizes were equal to 2,247 T-RFs per restriction endonuclease profile (2,247 corresponds to the lowest common denominator of T-RFs within the size range from 50 to 500 bp found by MseI).
. Menhinick's index approaches zero when there is a high number of individuals but few OTUs and, like Margalef's index, approaches a maximum value when the number of OTUs is equal to the number of individuals. D1 and D2 are sensitive to variations in sample size (26); therefore, data were normalized by randomly removing T-RFs from each restriction endonuclease profile until all sample sizes were equal to 2,247 T-RFs per restriction endonuclease profile (2,247 corresponds to the lowest common denominator of T-RFs within the size range from 50 to 500 bp found by MseI).
Iteration detection of restriction endonuclease fidelity.
Computer simulations were performed by repeated (n = 100) random samplings without replacement from the derived T-RF database for each restriction endonuclease. The simulation was designed to obtain and analyze model communities with fixed SSU rDNA richness values set at 1, 5, 10, 50, and 100 members. Community detection values were expressed as the probability of detecting a T-RF with a unique size or OTU within the range from 50 to 500 bp. To determine overall T-RFLP analysis efficacy, multiple restriction endonuclease profiles were combined for a single community. The community detection values were randomly sorted for each of the 100 independent samplings at each richness value. The average maximum community detection values were chosen for successive random restriction endonuclease selections.
RESULTS
The 4,603 T-RFLP-compatible sequences used in this analysis (31% of RDP, version 8.1) were uniformly distributed throughout the domain Bacteria (Table 2). The richness and density indices evaluated from those sequences showed that the restriction endonucleases used in this study have a high degree of variability in the ability to differentiate OTUs from the total T-RF data (Table 1). Hpy188III (rank, 1) and HhaI (rank, 2) were able to resolve the greatest number of OTUs (571 and 563 OTUs, respectively) while DdeI (rank, 17) and Sau96I (rank, 18) distinguished less than 300 OTUs (Table 1).
TABLE 2.
Phylogenetic distribution of the 4,603 RDP (version 8.1) sequences having primer sites matching the primers included in this study
| Major taxon | No. of sequences (% coverage) |
|---|---|
| Bacteria | |
| Proteobacteria | |
| α subdivision | 631 (32) |
| β subdivision | 290 (27) |
| γ subdivision | 824 (28) |
| δ subdivision | 168 (31) |
| ɛ subdivision | 95 (30) |
| High G+C content, gram positive | 926 (40) |
| Bacillus subdivision | 476 (38) |
| Other gram positive | 429 (31) |
| Flexibacter-Cytophaga-Bacteroides | 132 (17) |
| Cyanobacteria and chloroplasts | 150 (29) |
| Spirochetes | 144 (22) |
| Others | 338 (26) |
| Total | 4,603 (31) |
When T-RFs whose lengths were outside the range from 50 to 500 bp were removed (in an effort to represent the realistic resolving power of capillary and gel electrophoresis technology), the number of T-RFs associated with each restriction endonuclease was no longer the same for 4,603 sequences, and a different hierarchy of restriction endonucleases for detecting OTUs was found. BstUI ranked highest among the restriction endonucleases generating T-RFs within the range from 50 to 500 bp (Table 1). The disparity in rank after removal of T-RFs outside the desired range (50 to 500 bp) was mainly due to the presence of highly conserved cut sites. For example, BfaI has two highly conserved terminal cut sites in the size ranges from 570 to 601 and 745 to 760 bp. The resulting exclusion of 829 (18%) SSU rDNA T-RFs (data not shown) thereby decreased the rank of BfaI from 5th to 12th. Conversely, increases in rank (e.g., MspI changed from 11th to 1st) occurred when the majority of T-RFs for a restriction endonuclease were within the specified range from 50 to 500 bp (see reference 33 for a web-based tool to sort T-RFs by size).
Both the number of OTUs in the range from 50 to 500 bp (i.e., apparent richness) and density indices showed that MspI, Hpy188III, Tsp509I, and Hpy188I had the highest resolving capacities (Table 1). To remove the effect of unequal sample sizes on evaluating density indices, amplicons were randomly removed until the sample size for all communities was 2,247 (the lowest common denominator for T-RFs cut in the range from 50 to 500 bp by MseI). Because the effect of randomly removing data is not measurable, we used rarefaction analysis to compare communities with unequal sample sizes (19). The rarefaction curves agreed with the Margalef and Menhinick indices, showing that the same restriction endonucleases were the top performers in terms of the ability to identify the greatest number of OTUs (data not shown).
When 100 random communities were assembled at discrete richness values (i.e., 1, 5, 10, 50, and 100), BstUI outperformed (in terms of the number of OTUs detected) other restriction endonucleases at low richness values (Fig. 1). When the community richness value was set at 1, BstUI was able to detect the single population 98% of the time, while MseI performed the poorest by detecting the population in only 50% of the communities. When the richness value was increased to 5 and 10, a similar, yet declining resolution continued, with BstUI still outperforming the other enzymes. Once the T-RF richness value was increased to 50, all restriction endonucleases detected fewer than 71% of the OTUs, but there were seven restriction endonucleases (BstUI, DdeI, Sau96I, MspI, HinfI, HaeIII, and BslI) that still outperformed the others. When the T-RF richness value was set at 100, all restriction endonucleases performed uniformly poorly by detecting from 58% (MspI) to 35% (MseI) of the sequence variants. This decrease in fidelity is explained by an increasing frequency of identical T-RF sizes within a single community.
FIG. 1.

Percentage of OTUs detected for each restriction endonuclease at five discrete richness values (1, 5, 10, 50, and 100). The enzymes are arranged from left to right in decreasing order of the percentage of OTUs detected at a community richness value of 1. The error bars indicate standard errors of the means.
When restriction endonucleases were chosen in a random, sequential series, a plot was generated to characterize the number of enzymes needed to increase the detection of OTUs (Fig. 2). At a richness value of 1, the probability of detecting a T-RF reached 100% after five restriction endonucleases were used. At richness values of 5 and 10, similar curves were generated, in which the probability of detecting OTUs increased rapidly until after the use of three to five enzymes, at which point the probability of increasing the detection rose gradually. At richness values of 50 and 100, the detection ability only gradually increased to maximum values of 74 and 60%, respectively. The fraction of OTUs detected for each richness profile only gradually increased after four successive restriction endonucleases were used (Fig. 2).
FIG. 2.

Percentages of OTUs detected by randomly selecting from the pool of 18 restriction endonucleases at five discrete richness values (1, 5, 10, 50, and 100). The error bars indicate standard errors of the means. Lines are drawn between points to illustrate trends.
A second simulation was performed by using the subset of four restriction endonucleases (BstUI, DdeI, Sau96I, and MspI) that had the highest fidelity for detecting single-population communities (Fig. 1). The second simulation was analyzed at richness values of 1, 5, and 10 because all restriction endonucleases had performed poorly at richness values of 50 and 100. At a richness value of 1, the probability of detecting the single population reached 100% after only two restriction endonucleases were used. At richness values of 5 and 10, the top four performing restriction endonucleases were able to detect 98.8% ± 0.48% and 94% ± 0.72%, respectively (Fig. 3).
FIG. 3.

Percentages of OTUs detected by randomly selecting from a pool of four restriction endonucleases with the highest fidelity (BstUI, DdeI, Sau96I, and MspI) at three discrete richness values (1, 5, and 10). The error bars indicate standard errors of the means. Lines are drawn between points to illustrate trends.
DISCUSSION
The T-RFLP assay provides microbial ecologists with a rapid method for estimating community diversity and for screening samples to facilitate the prioritization of a sequencing effort. Community comparisons and other downstream analyses of T-RFLP data have been adopted via an assortment of statistical methods, such as similarity indices (8, 49), hierarchical clustering algorithms (4, 9, 10, 23, 35, 46), principal-component analyses (5, 8, 13), and self-organizing maps (8). In this study we focused on evaluating the ability of restriction endonucleases to resolve simulated community diversity by estimating confidence with respect to specific restriction endonucleases. Furthermore, we addressed whether T-RFLP analysis can be used as an accurate assay to characterize microbial diversity by combining data from multiple restriction endonuclease analyses for a single community. A single restriction endonuclease digest for a community with a richness value greater than 100 may reveal less than 50% of the simulated diversity (Fig. 2). For example, Dunbar et al. (9) found only 20 T-RFs in a soil community that contained 154 restriction fragment length polymorphism-derived clones. Consequently, the use of clustering, ordination, and neural networks may not give desirable results for high-diversity community comparisons. In our lab, we routinely compile the results from four to eight restriction endonuclease digests for a single community, yet we rarely observe useful similarities when we perform cluster analysis on communities with high levels of diversity. However, identifying dominant populations or performing statistical analyses on communities with low diversity remains a robust technique when T-RFLP data are utilized. In a recent study the investigators concluded that dominant populations can be detected by using T-RFLP analysis as a tool for precise quantification of the PCR product pool along with the capability for potential PCR bias detection (27).
The limitations of electrophoresis technology for accurately and precisely determining sizes of fragments within specific size ranges are well documented (15, 22, 24, 25, 28, 31, 47). The velocity at which a DNA fragment moves through a sieving matrix, such as agarose or polyacrylamide, is not linearly correlated with size. Small DNA fragments exhibit a high degree of separation as they travel rapidly through the matrix, which allows a high degree of precision in determining sizes. Unfortunately, there are a number of problems with including small fragments (<50 bp) in T-RFLP analysis, including the loss of small DNA fragments associated with the purification of samples, the unknown effects of Brownian motion, and the existence of residual PCR artifacts that may result in anomalous data (e.g., primer dimers). Because the migration time interval (or distance) between fragments decreases as the DNA fragment size increases, there is a maximum size (in base pairs) at which the resolution of DNA fragments having unique sizes is no longer possible. Consequently, the inclusion of large T-RFs (>500 bp) for data analysis is not recommended for fragment-analyzing technology. The advent of combined technologies that include pulsed-field gel electrophoresis (45) may eventually lead to higher-precision sizing and resolution of large DNA fragments for T-RFLP analysis.
The resolvability of a restriction endonuclease is its potential to detect OTUs from a set of sequence variants based on T-RF size distributions. Resolution can be directly analyzed by the use of diversity measurements (e.g., density indices, rarefaction curves). The fidelity of a restriction endonuclease is a more accurate measure and includes constraints involved in the T-RFLP assay. Fidelity can only be measured by simulation modeling because specific values of constraints are unknown. In this study, the ranking of restriction endonucleases based on the number of T-RFs in the size range from 50 to 500 bp was more important than the resolving ability in determining restriction endonuclease fidelity (compare rank values in Table 1 with Fig. 1). If fragment-analyzing technology included a larger range of T-RF sizes (e.g., 50 to 1,500 bp), resolvability complemented with a suitable PCR primer set would most likely be the best proxy for determining restriction endonuclease fidelity.
A random selection of sequences from the RDP database is not likely to be representative of communities found in nature due to the bias of clinical and repetitive entries found in the database (e.g., there are ∼45 SSU rRNA sequences from E. coli, 4 of which were identical in RDP, release 8.1). Currently, there are no methods used in molecular microbial ecology that accurately reflect a complete community found in nature. Consequently, we have no accurate measure for what a natural microbial community is in its entirety. Our results, therefore, show a lower, albeit more conservative, threshold for the T-RFLP range of detection.
Selection of appropriate primers for PCR-based microbial ecological studies is an ongoing task. The appropriate weighting of parameters that describe a suitable primer (e.g., melting temperature, level of similarity) is not yet agreed upon and must be determined empirically. We chose a previously tested degenerate primer set (11, 37) based on its ability to detect the greatest percentage of bacterial sequences from the RDP database compared with the abilities of other primer sets (data not shown). A further prerequisite for our selection of primers was to use bacterial primers spanning the majority of the SSU rDNA. The results of our experiment would not significantly vary if alternative terminal primers close to the 5′ end of the gene were selected, nor would they likely change dramatically if the reverse (i.e., 3′) primers were labeled as this would simply act just like another, albeit less informative, restriction enzyme treatment.
This study demonstrated that the type and number of restriction endonucleases are important parameters when an accurate representation of the diversity of a microbial community is desired. The specific restriction enzymes used may have to be empirically determined, especially in specialized habitats represented by limited numbers of phylotypes. However, we maintain that this modeling effort represents a good first-order approximation for choosing restriction enzyme treatment parameters for many types of environmental samples. The T-RFLP technique is likely to be a very valuable screening tool when spatiotemporal changes in natural communities with relatively low to intermediate species richness are studied. This technique may not be an adequate tool for characterizing complex microbial populations.
Acknowledgments
Special appreciation goes to the people at the RDP for their continued efforts in curatorship and making sequence data available to everyone. We also thank Emily Peele and Robin Matthews for critically reviewing the manuscript.
This research was supported in part by the Bureau of Faculty Research at Western Washington University and by the Washington State Sea Grant Program through project number R/B-34.
REFERENCES
- 1.Acinas, S., F. Rodriguez-Valera, and C. Pedros-Alio. 1997. Spatial and temporal variation in marine bacterioplankton diversity as shown by RFLP fingerprinting of PCR amplified 16S rDNA. FEMS Microbiol. Ecol. 24:27-40. [Google Scholar]
- 2.Amann, R. I., W. Ludwig, and K. Schleifer. 1995. Phylogenetic identification and in situ detection of individual cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avannis-Aghajani, E., K. Jones, D. Chapman, and C. Brunk. 1994. A molecular technique for identification of bacteria using small subunit ribosomal RNA sequences. BioTechniques 17:144-149. [PubMed] [Google Scholar]
- 4.Bruce, K. D., and M. R. Hughes. 2000. Terminal restriction fragment length polymorphism monitoring of genes amplified directly from bacterial communities in soils and sediments. Mol. Biotechnol. 16:261-269. [DOI] [PubMed] [Google Scholar]
- 5.Clement, B. G., L. E. Kehl, K. L. DeBord, and C. L. Kitts. 1998. Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities. J. Microbiol. Methods 31:135-142. [Google Scholar]
- 6.Cole, J. R., B. Chai, T. L. Marsh, R. J. Farris, Q. Wang, S. A. Kulam, S. Chandra, D. M. McGarrell, T. M. Schmidt, G. M. Garrity, and J. M. Tiedje. 2003. The ribosomal database project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delong, E. F., D. G. Franks, and A. L. Alldredge. 1993. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 38:924-934. [Google Scholar]
- 8.Dollhopf, S. L., S. A. Hashsham, and J. M. Tiedje. 2001. Interpreting 16S rDNA T-RFLP data: application of self-organizing maps and principal components analysis to describe community dynamics and convergence. Microb. Ecol. 42:495-505. [DOI] [PubMed] [Google Scholar]
- 9.Dunbar, J., L. O. Ticknor, and C. R. Kuske. 2000. Assessment of microbial diversity in four southwestern United States soils by 16S rRNA gene terminal restriction fragment length analysis. Appl. Environ. Microbiol. 66:2943-2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunbar, J., L. O. Ticknor, and C. R. Kuske. 2001. Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16S rRNA genes from bacterial communities. Appl. Environ. Microbiol. 67:190-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emerson, D., and C. L. Moyer. 2002. Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi Seamount hydrothermal vents and play a major role in Fe oxide deposition. Appl. Environ. Microbiol. 68:3085-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferris, M. J., and D. M. Ward. 1997. Seasonal distributions of dominant 16S rRNA-defined populations in a hot spring microbial mat examined by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 63:1375-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franklin, R. B., J. L. Garland, C. H. Bolster, and A. L. Mills. 2001. Impact of dilution on microbial community structure and functional potential: comparison of numerical simulations and batch culture experiments. Appl. Environ. Microbiol. 67:702-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giovannoni, S. J., T. B. Britschgi, C. L. Moyer, and K. G. Field. 1990. Genetic diversity of Sargasso Sea bacterioplankton. Nature 345:60-63. [DOI] [PubMed] [Google Scholar]
- 15.Grossman, P. D., S. Menchen, and D. Hershey. 1992. Quantitative analysis of DNA-sequencing electrophoresis. Genet. Anal. Tech. Appl. 9:9-16. [DOI] [PubMed] [Google Scholar]
- 16.Head, I. M., J. R. Saunders, and R. W. Pickup. 1998. Microbial evolution, diversity, and ecology: a decade of ribosomal RNA analysis of uncultivated microorganisms. Microb. Ecol. 35:1-21. [DOI] [PubMed] [Google Scholar]
- 17.Heyndrickx, M., L. Vauterin, P. Vandamme, K. Kersters, and P. De Vos. 1996. Applicability of combined amplified ribosomal DNA restriction analysis (ARDRA) patterns in bacterial phylogeny and taxonomy. J. Microbiol. Methods 26:247-259. [Google Scholar]
- 18.Horz, H., M. T. Yimga, and W. Liesack. 2001. Detection of methanotroph diversity on roots of submerged rice plants by molecular retrieval of pmoA, mmoX, mxaF, and 16S rRNA and ribosomal DNA, including pmoA-based terminal restriction fragment length polymorphism profiling. Appl. Environ. Microbiol. 67:4177-4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurlbert, S. H. 1971. The nonconcept of species diversity: a critique and alternative parameters. Ecology 52:577-586. [DOI] [PubMed] [Google Scholar]
- 20.Jeanthon, C. 2000. Molecular ecology of hydrothermal vent microbial communities. Antonie Leeuwenhoek 77:117-133. [DOI] [PubMed] [Google Scholar]
- 21.Kitts, C. L. 2001. Terminal-restriction fragment patterns: a tool for comparing microbial communities and assessing community dynamics. Curr. Issues Intest. Microbiol. 2:17-25. [PubMed] [Google Scholar]
- 22.Lerman, L. S., and H. L. Frisch. 1982. Why does the electrophoretic mobility of DNA in gels vary with the length of the molecule? Biopolymers 21:995-997. [DOI] [PubMed] [Google Scholar]
- 23.Liu, W., T. L. Marsh, H. Cheng, and L. J. Forney. 1997. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microbiol. 63:4516-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luckey, J. A., H. Drossman, A. J. Kostichka, D. A. Mead, J. D'Cunha, T. B. Norris, and L. M. Smith. 1990. High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 18:4417-4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luckey, J. A., T. B. Norris, and L. M. Smith. 1993. Analysis of resolution in DNA sequencing by capillary gel electrophoresis. J. Phys. Chem. 97:3067-3075. [Google Scholar]
- 26.Ludwig, J. A., and J. F. Reynolds. 1988. Statistical ecology. John Wiley & Sons, New York, N.Y.
- 27.Lueders, T., and M. W. Friedrich. 2003. Evaluation of PCR amplification bias by terminal restriction fragment polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl. Environ. Microbiol. 69:320-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lumpkin, O. J., P. Déjardin, and B. H. Zimm. 1985. Theory of gel electrophoresis of DNA. Biopolymers 24:1573-1593. [DOI] [PubMed] [Google Scholar]
- 29.Lynch, K. S. 2000. Bacterial community structure and phylogenetic diversity of hydrothermal vents at Axial Volcano, Juan de Fuca Ridge. M.S. thesis. Western Washington University, Bellingham.
- 30.Maidak, B. L., J. R. Cole, T. G. Lilburn, C. T. Parker, Jr., P. R. Saxman, R. J. Farris, G. M. Garrity, G. J. Olsen, T. M. Schmidt, and J. M. Tiedje. 2001. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 29:173-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manabe, T., N. Chen, and S. Terabe. 1994. Effects of linear polyacrylamide concentrations and applied voltages on the separation of oligonucleotides and DNA sequencing fragments by capillary electrophoresis. Anal. Chem. 66:4243-4252. [DOI] [PubMed] [Google Scholar]
- 32.Margalef, R. 1958. Information theory in ecology. Gen. Syst. 3:36-71. [Google Scholar]
- 33.Marsh, T. L., P. Saxman, J. Cole, and J. Tiedje. 2000. Terminal restriction fragment length polymorphism analysis program, a web-based research tool for microbial community analysis. Appl. Environ. Microbiol. 66:3616-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menhinick, E. P. 1964. A comparison of some species-individual diversity indices applies to samples of field insects. Ecology 45:859-861. [Google Scholar]
- 35.Moeseneder, M. M., J. M. Arrieta, G. Muyzer, C. Winter, and G. J. Herndl. 1999. Optimization of terminal-restriction fragment length polymorphism analysis for complex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 65:3518-3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moeseneder, M. M., C. Winter, J. M. Arrieta, and G. J. Herndl. 2001. Terminal-restriction fragment length polymorphism (T-RFLP) screening of a marine archaeal clone library to determine the different phylotypes. J. Microbiol. Methods 44:159-172. [DOI] [PubMed] [Google Scholar]
- 37.Moyer, C. L., F. C. Dobbs, and D. M. Karl. 1994. Estimation of diversity and community structure through restriction fragment length polymorphism distribution analysis of bacterial 16S rRNA genes from a microbial mat community at an active, hydrothermal vent system, Loihi Seamount, Hawaii. Appl. Environ. Microbiol. 60:871-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moyer, C. L., J. M. Tiedje, F. C. Dobbs, and D. M. Karl. 1996. A computer-simulated restriction fragment length polymorphism analysis of bacterial small-subunit rRNA genes: efficacy of selected tetrameric restriction enzymes for studies of microbial diversity in nature. Appl. Environ. Microbiol. 62:2501-2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moyer, C. L. 2001. Molecular phylogeny: applications and implications for marine microbiology. Methods Microbiol. 30:375-394. [Google Scholar]
- 40.Muyzer, G. 1999. DGGE/TGGE: a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 2:317-322. [DOI] [PubMed] [Google Scholar]
- 41.Nei, M., and W.-H. Li. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76:5269-5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborn, A. M., E. R. Moore, and K. N. Timmis. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2:39-50. [DOI] [PubMed] [Google Scholar]
- 43.Pace, N. R. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734-740. [DOI] [PubMed] [Google Scholar]
- 44.Rath, J., K. Y. Wu, G. J. Herndl, and E. F. Delong. 1998. High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 14:261-269. [Google Scholar]
- 45.Schwartz, D. C., and C. R. Cantor. 1984. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37:67-75. [DOI] [PubMed] [Google Scholar]
- 46.Sessitsch, A., A. Weilharter, M. H. Gerzabek, H. Kirchmann, and E. Kandeler. 2001. Microbial population structures in soil particle size fractions of a long-term fertilizer field experiment. Appl. Environ. Microbiol. 67:4215-4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swerdlow, H., J. Z. Zhang, D. Y. Chen, H. R. Harke, R. Grey, S. Wu, and N. J. Dovichi. 1991. Three DNA sequencing methods using capillary gel electrophoresis and laser-induced fluorescence. Anal. Chem. 63:2835-2841. [DOI] [PubMed] [Google Scholar]
- 48.Vaneechoutte, M., and R. Rossau. 1992. Rapid identification of bacteria of the Comamonadaceae with amplified ribosomal DNA-restriction analysis (ARDRA). FEMS Microbiol. Lett. 93:227-234. [DOI] [PubMed] [Google Scholar]
- 49.Watts, J. E. M., Q. Wu, S. B. Schreier, H. D. May, and K. R. Sowers. 2001. Comparative analysis of polychlorinated biphenyl-dechlorinating communities in enrichment cultures using three different molecular screening techniques. Environ. Microbiol. 3:710-719. [DOI] [PubMed] [Google Scholar]
- 50.Weidner, S., W. Arnold, and A. Puhler. 1996. Diversity of uncultured microorganisms associated with the seagrass Halophila stipulacea estimated by restriction fragment length polymorphism analysis of PCR-amplified 16S rRNA genes. Appl. Environ. Microbiol. 62:766-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woese, C. R. 1987. Bacterial evolution. Microbiol. Rev. 51:221-271. [DOI] [PMC free article] [PubMed] [Google Scholar]