

Abstract
The human opportunistic pathogen Pseudomonas aeruginosa causes a variety of infections in immunocompromised hosts and in individuals with cystic fibrosis. A knockout mutation in the polyphosphate kinase (ppk) gene, encoding PPK responsible for the synthesis of inorganic polyphosphate from ATP, renders P. aeruginosa cells unable to form a thick and differentiated biofilm. The mutant is aberrant in quorum sensing and responses in that production of the quorum-sensing controlled virulence factors elastase and rhamnolipid are severely reduced. In a burned-mouse pathogenesis model, the virulence of the mutant is greatly reduced with severe defects in the colonization of mouse tissues. The conservation of PPK among many bacterial pathogens and its absence in eukaryotes suggest that PPK might be an attractive target for antimicrobial drugs.
Inorganic polyphosphate (poly P) is a linear polymer of hundreds of orthophosphate (Pi) residues linked by ATP-like, high-energy, phosphoanhydride bonds and found in all microbes, fungi, plants, and animals (1). The enzyme responsible for the synthesis of poly P in bacteria is poly P kinase (PPK), which polymerizes the terminal phosphate of ATP into a poly P chain (2). One of the functions of poly P in Escherichia coli is a regulatory role inferred from the inability of the ppk mutant to adapt to nutritional stringencies and environmental stresses and to survive in the stationary phase of growth (3, 4). These defects of the ppk mutant have been attributed partly to the failure to express the rpoS gene that encodes σs, the stationary phase σ factor (5).
PPK is highly conserved in many bacterial species, both Gram-positive and Gram-negative, including some major pathogens (6). Inasmuch as some virulence factors are expressed in the stationary phase(7, 8), we prepared ppk knockouts in several pathogens including Pseudomonas aeruginosa, Klebsiella pneumoniae, Vibrio cholerae, Salmonella typhimurium, Salmonella dublin, and Helicobacter pylori to examine their phenotypic features and dependence on PPK for virulence (9). The ppk mutants of these pathogens are moderately defective in flagella-mediated swimming on semisolid agar plates (9). In addition, the P. aeruginosa ppk mutant is defective in a newly discovered swarming motility dependent on flagella and in a previously characterized twitching motility that depends on type IV pili (10).
Biofilms are sessile microbial communities formation of which are initiated by surface attachment of individual planktonic bacteria, followed by cell–cell interactions that develop into growing colonies in an elaborate three-dimensional structure (11). Highly differentiated mushroom- and pillar-like biofilm structures, consisting largely of mucoid exopolysaccharide, are bathed by water-filled channels. Formation of biofilms is a multistep developmental process over a period of several hours (12). It resembles spore formation in Bacillus subtilis, the multicellular fruiting body development in Myxococcus xanthus, and stalked-cell formation by Caulobacter crescentus (13). Recent genetic screens of biofilm-defective mutants in E. coli, P. aeruginosa, and V. cholerae have revealed that initial surface interaction followed by microcolony formation and enlargement are mediated by force-generating organelles: flagella and pili (14–16). Exopolysaccharide production is necessary to stabilize the pillars of the biofilm (16, 17). Finally, intercellular communication through the diffusing signaling molecules determines the ultimate three-dimensional architecture of the mature biofilms (18).
Bacteria communicate their cell density by quorum sensing to coordinate the expression of particular genes (19). Among quorum-signaling molecules or autoinducers (AIs), homoserine lactones (HSLs) control expression of extracellular virulence factors (e.g., toxins, elastases, and proteases) and small-molecule, secondary metabolites (e.g., rhamnolipid, phenazine, and cyanide) in P. aeruginosa (20). Two distinctive, semiindependent quorum-sensing systems, the lasR–lasI and the rhlR–rhlI regulons, have been characterized in P. aeruginosa (21–31). In the first system, the lasI gene product catalyzes the formation of the AI-1 signal, N-(3-oxododecanoyl)-L-HSL, which interacts with LasR, a transcriptional regulator, to activate a number of genes including lasI itself, the elastase genes lasA and lasB, and the rhlR gene of the second system (21–23, 29–31). The rhlI product directs the synthesis of the AI-2 signal, N-butyryl-L-HSL, required for the activation of several genes including rhlI itself, the rhlA gene encoding rhamnosyltransferase for rhamnolipid biosynthesis, and the rpoS gene by RhlR complexed with AI-2 (24–31). Of these signals, only AI-1 is required for the normal maturation of P. aeruginosa biofilms (18). With regard to twitching, the AI-2 signal is absolutely necessary whereas the AI-1 contribution is only partial (35% residual twitching in the lasI mutant) (32).
P. aeruginosa is a ubiquitous Gram-negative bacterium commonly found in soil, water, and plants, and is an opportunistic human pathogen that causes serious infections in cystic fibrosis patients, in immunocompromised hosts including patients with cancer, HIV infections, and in severe burns and wounds (20). P. aeruginosa also has been reported to cause diseases in plants, insects, worms and a variety of vertebrates (33, 34). The success of P. aeruginosa in diverse environments is attributed to its impressive arsenal of virulence factors, which include multiple cell-associated factors, i.e., alginate (an exopolysaccharide), lipopolysaccharide, flagella and pili, and secreted virulence factors, including toxins, elastases, protease, phospholipase, as well as small molecules that include phenazines, rhamnolipid, and cyanide (20, 33). The virulence of P. aeruginosa (as well as the roles of specific factors in its virulence) has been examined by using different animal as well as plant and invertebrate models (35–40). Among the animal models, the burned-mouse model recently has been successfully used to demonstrate essential roles of different quorum-sensing components (41).
We reported earlier that poly P and PPK are required for three different forms of motility in P. aeruginosa (9, 10). In this report, we demonstrate that they are essential for quorum sensing and virulence of this clinically important pathogen. This result identifies PPK as a target for the development of a new class of antibacterial drugs.
Materials and Methods
Bacterial Strains and Plasmids.
P. aeruginosa strains used in this study were PAO1 (wild type; WT) and PAOM5 [ppk∷tetracycline resistance (TcR)] (9, 10). Plasmid pHEPAK11 for complementation in the PAOM5 strain was constructed as follows. The P. aeruginosa ppk gene was amplified from the plasmid pPPK02F (42) by PCR with the forward primer 5′-gcgAAGCTTCCCTCGGGAAGATGAATGAATACG-3′ (gcg clamp in lowercase and HindIII site in bold, followed by a 24-mer stretch of DNA starting at 154 bp upstream of the GTG translational start codon of the ppk gene (43) and the reverse primer 5′-gcgGATATCTCAACGTGCGGTAAGCACCGG-3′ (gcg clamp in lowercase, EcoRV site in bold and followed by a 21-mer stretch of DNA starting at the TGA stop codon of the ppk gene). The 2.23-kb PCR product was cloned into the HindIII/SmaI site of the E. coli–P. aeruginosa shuttle vector pMMB66HE (44), which places the ppk gene under the control of isopropyl β-d-thiogalactoside-inducible tac promoter. After checking in E. coli DH5α for the overexpression of PPK activity, this plasmid was electroporated into the P. aeruginosa ppk mutant. Resistance to carbenicillin is conferred by pHEPAK11 both in E. coli and in P. aeruginosa.
Biofilm Assays.
Static biofilms.
Cells were grown in M63 minimal media supplemented with 0.2% glucose, 1 mM MgSO4, and 0.5% casamino acids (45) at 30°C for static biofilm experiments. (i) Quantitation of biofilm bacteria. Experiments were performed as described earlier (45). The strains were cultured in separate wells of a polystyrene microtiter dish followed by staining with crystal violet; the cell-attached dye was solubilized with ethanol and measured at OD595. (ii) Epifluorescence and scanning confocal laser microscopy (SCLM). The strains harboring the green fluorescent protein (GFP) expression vector pMRP9–1 (18) were grown for 18 h in glass chambers containing borosilicate coverglass (no. 1) bottoms (chambered coverglass systems, Nalge Nunc). The chambers were emptied, washed with water, and examined with a scanning confocal laser microscope (MultiProbe 2010, Molecular Dynamics) using 488- and 510-nm excitation and emission wave lengths, respectively.
Continuous flow-cell biofilm.
Flow-cell biofilm experiments were performed in EPRI medium (0.005% sodium lactate/0.005% sodium succinate/0.005% ammonium nitrate/0.00019% KH2PO4/0.00063% K2HPO4, pH 7.0/0.001% Hunter salts/0.001% L-histidine) (18) supplemented with 0.1% dextrose (pH 7.0) in a bioreactor of the once-through, continuous-culture type (18). Cell clusters are defined as assemblages of bacteria greater than 10 μm in thickness. Cell-cluster thickness determinations were made by using transmitted light microscopy to determine the base of the biofilm cell cluster at the substratum and the apex of the biofilm cell cluster at the bulk-liquid interface farthest from the substratum. Cell cluster surface area coverage measured the total area in a microscope field occupied by cell clusters.
Extraction and Bioassays of AI-1 and AI-2.
Extraction. P. aeruginosa cultures were grown to early stationary phase (OD600 of 1.5) in peptone trypticase soy broth at 37°C with shaking. AI-1 and AI-2 were extracted from the culture supernatants of the ppk mutant with ethyl acetate (23).
Bioassays.
Bioassays were performed in E. coli reporter strains grown in modified A medium. AI-1 bioassays were performed with E. coli MG4 λI14 harboring a lasI∷lacZ fusion in a monolysogen (46). AI-2 bioassays were performed in E. coli DH5α harboring the pECP61.5 plasmid containing an rhlA-lacZ fusion construct (47). For AI-1 bioassay, a 1-ml overnight culture diluted 1:100 was mixed with the sample and grown for 3–4 h at 37°C. For AI-2 bioassay, overnight cultures were diluted 1:100 and grown at 37°C with shaking to an OD600 of 0.3; 1-ml samples were then further grown 90 min with 1 mM isopropyl β-d-thiogalactoside in the presence of AI-2. Comparison of the β-galactosidase values obtained with the extracted AIs against those of standard curves plotted with the synthetic AIs allowed the estimation of AI content in each sample.
Elastase and Rhamnolipid Assays.
For elastase activity measurements, cells were grown in peptone trypticase soy broth at 37°C for 20 h with shaking; elastase activity was measured by the elastin Congo red assay (47). For the measurement of rhamnolipid, cells were grown in modified Guerra-Santos (GS) medium at 37°C for 80 h with shaking; rhamnose content was determined by oricinol assays and compared to rhamnose standards (47).
β-Galactosidase Measurements.
Activity was measured as described (48). E. coli was grown in modified A medium with shaking at 37°C to an OD600 of 0.5–0.8. P. aeruginsoa was grown in Luria–Bertani medium at 30°C with shaking for 20 h. Strains PAO1 and PAOM5 harbored either plasmid pTS400 (lasB-lacZ) or pECP60 (rhlA-lacZ) (47).
Virulence Assays.
Inoculum preparation.
P. aeruginosa inocula for virulence studies were prepared as described earlier (41, 49). Overnight cultures were diluted 1:100 into fresh Luria–Bertani medium and incubated at 37°C for 4 h (OD540 = 0.9–1.0.). A sample (100 μl) from each culture was pelleted, washed in PBS, and serially diluted. An aliquot (100 μl) of a 10−5 dilution was injected containing approximately 200–300 colony-forming units (CFUs) of P. aeruginosa that produces 94–100% lethality by 48 h postburn (41, 49).
Burned Mouse Model.
Virulence experiments were conducted with adult female ND4 Swiss Webster mice weighing 20–24 g with burn lesions as described earlier (41, 49). The mice were anesthetized by i.p. injection of 0.4 ml of 5 mg/ml Nembutal (5% sodium pentobarbital; Abbott). Fluid replacement therapy consisting of a s.c. injection of 0.8 ml 0.9% NaCl solution was administered immediately after the burn. Mice were inoculated s.c. with ≈200 CFU directly under the burn whereas control mice received 100 μl of sterile PBS solution. During recovery, the mice were observed under warming lights. Animals were treated humanely and in accordance with the protocol approved by the Animal Care and Use Committee at Texas Tech University Health Sciences Center in Lubbock, TX. Three groups of five mice each were burned and infected for each experiment. For in vivo lethality assay, mortality was recorded at 48-h postburn infection. For horizontal spread, mice were killed and two sections (≈5 × 5 mm each) of the burned skin were removed from each animal at 8- and 24-h postburn infection. One section was obtained from the inoculation site, whereas the other was obtained ≈1.5 cm distant. For systemic spread, livers and spleens of infected mice were collected at 24-h postburn infection.
CFU Determinations.
Mouse tissues were suspended in PBS and homogenized, and an aliquot of the homogenate was plated on Luria–Bertani agar plates to determine CFU/g of tissue (41, 49).
Results and Discussion
PPK is Essential for Biofilm Development.
In P. aeruginosa biofilm development, flagella-mediated swimming motility is needed for the initial attachment of individual cells to an abiotic surface followed by microcolony formation mediated by the type IV pili-mediated twitching motility (15). The subsequent development of elaborate three-dimensional structures requires the quorum-sensing lasR–lasI system (18). Inasmuch as the ppk mutant is defective in swimming, swarming, and twitching (9, 10), we measured the capacity of the mutant to attach to and form biofilms on an abiotic surface in a simple, static-culture system that is less dependent on attachment. As expected (15), the ppk mutant is moderately defective in attachment (at 8 h) to a polystyrene surface (Fig. 1A). But even past attachment (at 20 h), another defect (presumably maturation of biofilm) was apparent, which was restored to WT level by complementation with the ppk gene (Fig. 1B). These defects do not arise from growth impairment because the mutant had a growth rate indistinguishable from the WT (9). Thus, biofilm maturation as well as surface attachment is greatly affected in the ppk mutant.
Figure 1.
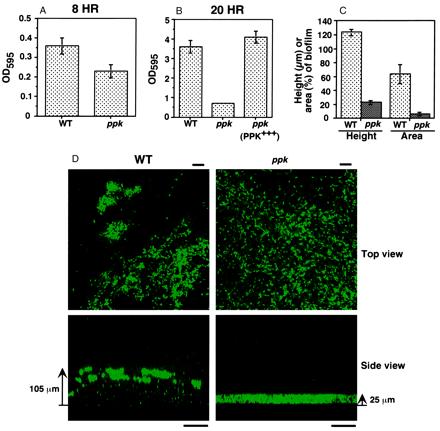
Biofilms of P. aeruginosa WT and ppk mutant strains in static cultures (A, B, and D) and flow-through continuous cultures (C). (A and B) Degree of surface attachment of bacteria at 8 h (A) and at 20 h (B) by staining with crystal violet (average ± SEM of four measurements). (C) Height of biofilms (in μm) (average ± SEM for 75 measurements in three separate experiments) and surface area coverage of biofilms (as percentage of total flow-cell area) (average ± SEM of 15 measurements in three separate experiments). (D) Epifluorescence and SCLM of static biofilms of the WT and ppk mutant containing the GFP expression vector pMRP9–1 (18). The top view epifluorescence micrographs were taken (with a X60 lens) at 5 μm from the substratum. Bar is 10 μm. The side views were acquired by SCLM (with a X20 lens). Bar is 50 μm. Bacterial cells contain GFP and the color intensity correlates with cell density. Strains: WT, PAO1; ppk, PAOM5; and ppk (PPK+++), PAOM5 containing pHEPAK11.
To compare the architecture of WT and ppk mutant biofilms, a plasmid with a gene encoding an enhanced GFP (18) enabled the viewing of static-culture biofilms by epifluorescence and SCLM (Fig. 1D). The side view of the 18-h static-biofilm acquired by SCLM revealed adherent clusters of WT cells; they appeared to be in loose aggregates with considerable intervening spaces between them. By contrast with the WT biofilm (105 ± 5 μm), the mutant biofilm was thin (25 ± 1 μm) and much more uniform. A top view generated by epifluorescence microscopy showed the WT cells in clusters as compared to a much more uniform distribution of the ppk mutant. The overall architecture of the 18-h static biofilm is very similar to that of a 2-wk flow-cell biofilm (18).
In a continuous-flow cell, the three-dimensional architecture of WT biofilms and that of the ppk mutant reached steady-state levels within 10 days, as expected (18). As with the mature static biofilms (Fig. 1D), the ppk mutant biofilm was only ≈20% of the WT thickness (Fig. 1C). The substratum surface area coverage by cell clusters of the mutant biofilm was only 10% (or less) of the WT. Microcolonies composed of groups of WT cells were separated by water channels, whereas the ppk mutant appeared to grow as a continuous sheet on the glass surface. Thus, for the differentiation of mature biofilms, viewed either in static or continuous-flow systems, the ppk mutant is profoundly deficient. This biofilm maturation defect of the ppk mutant is similar to the one that was seen in a lasI mutant deficient in the synthesis of AI-1 quorum-sensing molecule (18).
Effect of the ppk Mutation on the Synthesis of Autoinducers and Extracellular Virulence Factors.
Inasmuch as the ppk mutant is defective in three types of motility (9, 10), surface attachment and biofilm differentiation (Fig. 1), we determined the levels of quorum-sensing molecules AI-1 and AI-2 in the culture supernatant of the ppk mutant by bioassays using E. coli reporter strains (Fig. 2A). AI-1 and AI-2 levels in the ppk mutant were reduced to ≈50% those of the WT; complementation of the mutant with the ppk gene doubled the WT levels. Clearly, PPK modulates the synthesis of both AIs in P. aeruginosa.
Figure 2.
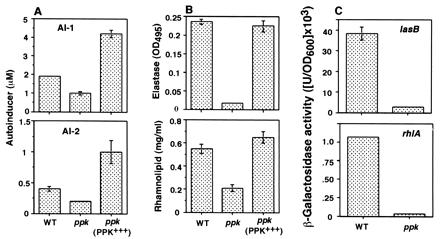
Quorum sensing in the P. aeruginosa ppk mutant. (A) Bioassays of AI-1 and AI-2 (average ± SEM of six measurements in three separate experiments). (B) Elastase activity and rhamnolipid amount (average ± SEM of three measurements) in P. aeruginosa culture supernatants determined by elastin Congo red and oricinol assays, respectively. (C) Expression of the lasB-lacZ and the rhlA-lacZ fusion constructs contained in plasmids in the ppk mutant. β-Galactosidase activity (average ± SEM of three measurements) was measured as described in Materials and Methods. Strains are as in Fig. 1.
Because the production of extracellular virulence factors including elastase are under quorum-sensing control (21, 22), we examined qualitatively the level of elastase activity on elastin agar plate as a guide to virulence factor production in the mutant. The activity was reduced in the mutant that could be complemented with the plasmid expressing the ppk gene (data not shown). The total elastase activity and the total rhamnolipid amount, determined quantitatively in the culture supernatant of the ppk mutant were reduced to 7% and 38% of the WT levels, respectively; complementation with ppk restored the WT levels (Fig. 2B). With respect to the quorum-sensing target gene lasB for the major elastase and rhlA for a rhamnosyltransferase required for rhamnolipid biosynthesis, their expression in the ppk mutant determined with lacZ fusion constructs, was reduced to 7% for lasB–lacZ whereas that of the rhlA-lacZ was reduced to 3% of the WT levels (Fig. 2C). These data suggest that PPK and/or poly P affects the quorum-sensing system in the synthesis of AIs and probably also in the formation of AI complexes with cognate regulatory proteins. Alternatively, the ppk mutation, at another level, may affect the transcriptional activation of downstream target genes in interactions with RNA polymerase and σ factors (5, 50). The drastic effects of the ppk mutation on lasB and rhlA expression might thus represent actions at more than one level.
PPK Is Necessary for Virulence in the Burned-Mouse Model.
The burned-mouse model (41, 49) was used to examine the effect of the ppk mutation on the pathogenesis of P. aeruginosa infections in burn wounds. The nonlethal burn injury is created by a thermal shock (with 90°C water for 10 s) to the shaved back of a mouse (≈15% of the body surface area). P. aeruginosa injected s.c. into the center of the burn wound proliferates and invades the underlying tissues. The infection spreads horizontally and systemically. In three separate experiments, only one mouse out of 19 survived inoculation with WT bacteria as compared to the survival of 14 out of 15 inoculated with the ppk mutant (Table 1). This result for the ppk mutant is the same as observed in the quorum-sensing mutant that lacks both AIs (41). Complementation of the ppk mutant bacteria with PPK raised the mortality of mice from 7% to 53%. This partial complementation may be due to gene dosage effects because overexpression of PPK in E. coli has been found to be lethal (N. N. Rao & A.K., unpublished result).
Table 1.
Lethality of the P. aeruginosa ppk mutant in a burned-mouse model
| Strain | Relevant genotype | Mortality, mice dead/total*
|
|||
|---|---|---|---|---|---|
| Exp. 1 | Exp. 2 | Exp. 3 | Average, % | ||
| PAO1 | WT | 7/7 | 5/6 | 6/6 | 94 |
| PAOM5 | Δppk∷TcR | 1/5 | 0/5 | 0/5 | 7 |
| PAOM5 (pHEPAK11) | Δppk∷TcR (PPK+++) | 2/5 | 3/5 | 3/5 | 53 |
Burned-mice were inoculated s.c. with ≈200 cells of each strain; mortality was checked after 48 h. Exp., experiment.
With regard to horizontal spread within the burned skin, the ppk mutant bacteria were completely absent at a distant site at 8 h, and a few were found at the inoculation site compared to the WT bacteria (Table 2). The levels of the mutant bacteria both at inoculation and distant sites at 24 h increased but remained low compared to the WT bacteria. Systemic spread of the ppk mutant to the liver and spleen at 24 h postinfection was <1% that of the WT (Table 2). The effects of the ppk mutation on both horizontal and systemic spreads are even more drastic than those observed with the quorum-sensing mutant deficient in both AI-1 and AI-2 production (41).
Table 2.
Horizontal spread and systemic infection of the P. aeruginosa ppk mutant in mouse tissues
| Strain | CFU/g tissue*
|
|||||
|---|---|---|---|---|---|---|
| Horizontal spread within skin
|
Systemic infection
|
|||||
| 8 h, Inoc.
|
8 h, Dist.
|
24 h, Inoc.
|
24 h, Dist.
|
Spleen
|
Liver
|
|
| ×104 | ×104 | ×107 | ×107 | ×103 | ×102 | |
| PAO1 (WT) | 180 ± 70 | 30 ± 7.6 | 180 ± 32 | 120 ± 28 | 460 ± 65 | 309 ± 61 |
| PAOM5 (ppk) | 4.4 ± 1.8 | 0 | 120 ± 57 | 39 ± 10 | 2 ± 1 | 1 ± 0.4 |
Mice were injected with ≈200 cells. CFUs were measured at 8 h and 24 h post-burn infection for horizontal spread within the skin and at 24 h for systemic infection in spleen and liver. Values represent averages ± SEM for 15 measurements in three separate experiments. Inoc., inoculation site; Dist., distant site (1.5 cm away).
Conclusion.
These studies demonstrate that PPK is essential in P. aeruginosa not only for various forms of motility (9, 10) but also for the development of biofilms, production of the virulence factors elastase and rhamnolipid, and for virulence in the burned-mouse pathogenesis model. All of these effects are likely exerted through a defect in quorum sensing and responses. Because formation of biofilms and their inherent resistance to antimicrobial agents are at the root of many persistent and chronic infections such as the lungs of cystic fibrosis patients (51), PPK qualifies as a therapeutic target to control biofilm infections by perturbing the integrity of the quorum sensing system. An antimicrobial drug targeted to PPK should enjoy a broad spectrum of activity and little toxicity, inasmuch as the enzyme has not been found in mammalian cells (52). As PPK is involved in cellular metabolism rather than in an essential function, drugs targeted to it might be less likely to provoke resistance (53). Because PPK is highly conserved in both Gram-positive and Gram-negative pathogens (6, 52), the inhibitors of PPK may block quorum-sensing at an upstream level, as opposed to the analogues of specific quorum-signaling molecules (54–56) in switching off virulence gene expression and thereby attenuating pathogenicity.
Acknowledgments
We thank S. L. Palmieri for performing epifluorescence and SCLM, T. Noguchi and T. Shiba for plasmid pPPK02F, E. P. Greenberg for plasmid pMRP9–1, K. Kreitz for technical assistance, and D. Kaiser, L. Bertsch, and C. Fraley for critical reading of the manuscript. Support was provided by grants to B.H.I and A.K. from the National Institutes of Health and by the Cystic Fibrosis Foundation to A.K.
Abbreviations
- poly P
inorganic polyphosphate
- PPK
polyphosphate kinase
- WT
wild type
- HSL
homoserine lactone
- AI
autoinducer
- CFU
colony-forming unit
- SCLM
scanning confocal laser microscopy
- GFP
green fluorescent protein
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170283397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170283397
References
- 1.Kulaev I S. The Biochemistry of Inorganic Polyphosphates. New York: Wiley; 1979. [DOI] [PubMed] [Google Scholar]
- 2.Ahn K, Kornberg A. J Biol Chem. 1990;265:11734–11739. [PubMed] [Google Scholar]
- 3.Rao N N, Kornberg A. J Bacteriol. 1996;178:1394–1400. doi: 10.1128/jb.178.5.1394-1400.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ault-Riché D, Fraley C F, Tzeng C-M, Kornberg A. J Bacteriol. 1998;180:1841–1847. doi: 10.1128/jb.180.7.1841-1847.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiba T, Tsutsumi K, Yano H, Ihara Y, Kameda A, Tanaka K, Takahashi H, Masanobu M, Rao N N, Kornberg A. Proc Natl Acad Sci USA. 1997;94:11210–11215. doi: 10.1073/pnas.94.21.11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tzeng C-M, Kornberg A. Mol Microbiol. 1998;29:381–382. doi: 10.1046/j.1365-2958.1998.00887.x. [DOI] [PubMed] [Google Scholar]
- 7.Loewen P C, Hengge-Aronis R. Annu Rev Microbiol. 1994;48:53–80. doi: 10.1146/annurev.mi.48.100194.000413. [DOI] [PubMed] [Google Scholar]
- 8.Spector M P. Adv Microb Physiol. 1998;40:233–279. doi: 10.1016/s0065-2911(08)60133-2. [DOI] [PubMed] [Google Scholar]
- 9.Rashid M H, Rao N N, Kornberg A. J Bacteriol. 2000;182:225–227. doi: 10.1128/jb.182.1.225-227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashid M H, Kornberg A. Proc Natl Acad Sci USA. 2000;97:4885–4890. doi: 10.1073/pnas.060030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costerton J W, Lewandowski Z, Caldwell D E, Korber D R, Lappin-Scott H M. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- 12.Kolter R. J Bacteriol. 2000;182:2675–2679. doi: 10.1128/jb.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Losick R, Kaiser D. Sci Am. 1997;276:68–73. doi: 10.1038/scientificamerican0297-68. [DOI] [PubMed] [Google Scholar]
- 14.Pratt L A, Kolter R. Mol Microbiol. 1998;30:285–293. doi: 10.1046/j.1365-2958.1998.01061.x. [DOI] [PubMed] [Google Scholar]
- 15.O'Toole G A, Kolter R. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 16.Watnick P I, Kolter R. Mol Microbiol. 1999;34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoyle B D, Williams L J, Costerton J W. Infect Immun. 1993;61:777–780. doi: 10.1128/iai.61.2.777-780.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies D G, Parsek M R, Pearson J P, Iglewski B H, Costerton J W, Greenberg E P. Science. 1998;280:295–298. doi: 10.1126/science.280.5361.295. [DOI] [PubMed] [Google Scholar]
- 19.Fuqua W C, Winans S C, Greenberg E P. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deldon C V, Iglewski B H. Emerg Infect Dis. 1998;4:551–560. doi: 10.3201/eid0404.980405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gambello M J, Iglewski B H. J Bacteriol. 1991;173:3000–3009. doi: 10.1128/jb.173.9.3000-3009.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Passador L, Cook J M, Gambello M J, Rust L, Iglewski B H. Science. 1993;260:1127–1130. doi: 10.1126/science.8493556. [DOI] [PubMed] [Google Scholar]
- 23.Pearson J P, Gray K M, Passador L, Tucker K D, Eberhard A, Iglewski B H, Greenberg E P. Proc Natl Acad Sci USA. 1994;91:197–201. doi: 10.1073/pnas.91.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ochsner U A, Koch A K, Fiechter A, Reiser J. J Bacteriol. 1994;176:2044–2054. doi: 10.1128/jb.176.7.2044-2054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latifi A, Winson K M, Foglino M, Bycroft B W, Stewart G S A B, Lazdunski A, Williams P. Mol Microbiol. 1995;17:333–344. doi: 10.1111/j.1365-2958.1995.mmi_17020333.x. [DOI] [PubMed] [Google Scholar]
- 26.Brint J M, Ohman D E. J Bacteriol. 1995;177:7155–7163. doi: 10.1128/jb.177.24.7155-7163.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson J P, Passador L, Iglewski B H, Greenberg E P. Proc Natl Acad Sci USA. 1995;92:1490–1494. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochsner U A, Reiser J. Proc Natl Acad Sci USA. 1995;92:6424–6428. doi: 10.1073/pnas.92.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pesci E C, Iglewski B H. Trends Microbiol. 1997;5:132–135. doi: 10.1016/S0966-842X(97)01008-1. [DOI] [PubMed] [Google Scholar]
- 30.Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. Mol Microbiol. 1996;21:1137–1146. doi: 10.1046/j.1365-2958.1996.00063.x. [DOI] [PubMed] [Google Scholar]
- 31.Pesci E C, Pearson J P, Seed P C, Iglewski B H. J Bacteriol. 1997;179:3127–3132. doi: 10.1128/jb.179.10.3127-3132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glessner A, Smith R S, Iglewski B H, Robinson J B. J Bacteriol. 1999;181:1623–1629. doi: 10.1128/jb.181.5.1623-1629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollack M. In: Principles and Practice of Infectious Diseases. 5th Ed. Mandell G L, Bennett J E, Dolin R, editors. New York: Churchill Livingstone; 1999. pp. 2310–2327. [Google Scholar]
- 34.Finlay B B. Cell. 1999;96:315–318. doi: 10.1016/s0092-8674(00)80544-9. [DOI] [PubMed] [Google Scholar]
- 35.Rahme, L. G., Stevens, E. J., Wolfort, S. F., Shao, J., Tompkins, R. G. & Ausubel, F. M. Science268, 1899–1902. [DOI] [PubMed]
- 36.Tan M-W, Mahajan-Miklos S, Ausubel F M. Proc Natl Acad Sci USA. 1999;96:715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahajan-Miklos S, Tan M-W, Rahme L G, Ausubel F M. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 38.Tang H B, DiMango E, Bryan R, Gambello M, Iglewski B H, Goldberg J, Prince A. Infect Immun. 1996;64:37–43. doi: 10.1128/iai.64.1.37-43.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, Prince A. Infect Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H, Kays M, Prince A. Infect Immun. 1995;63:1278–1285. doi: 10.1128/iai.63.4.1278-1285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rumbaugh K P, Griswold J A, Iglewski B H, Hamood A N. Infect Immun. 1999;67:5854–5862. doi: 10.1128/iai.67.11.5854-5862.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishige K, Kameda A, Noguchi T, Shiba T. DNA Res. 1998;5:157–162. doi: 10.1093/dnares/5.3.157. [DOI] [PubMed] [Google Scholar]
- 43.Zago A, Chugani S, Chakrabarty A M. Appl Environ Microbiol. 1999;65:2065–2071. doi: 10.1128/aem.65.5.2065-2071.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furste J P, Pansegran W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 45.O'Toole G A, Kolter R. Mol Microbiol. 1998;28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 46.Seed P C, Passador L, Iglewski B H. J Bacteriol. 1995;177:654–659. doi: 10.1128/jb.177.3.654-659.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearson J P, Pesci E C, Iglewski B H. J Bacteriol. 1997;179:5756–5767. doi: 10.1128/jb.179.18.5756-5767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 49.Rumbaugh K P, Griswold J A, Hamood A N. J Burn Care Rehab. 1999;20:42–49. doi: 10.1097/00004630-199901001-00008. [DOI] [PubMed] [Google Scholar]
- 50.Kusano S, Ishihama A. Genes Cells. 1997;2:433–441. doi: 10.1046/j.1365-2443.1997.13203301320330.x. [DOI] [PubMed] [Google Scholar]
- 51.Costerton J W, Stewart P S, Greenberg E P. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 52.Kornberg A, Rao N N, Ault-Riche D. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 53.McKenna N. Gen Eng News. 2000;20:1. , 38, 72. [Google Scholar]
- 54.Hartman G, Wise R. Lancet. 1998;351:848–849. doi: 10.1016/S0140-6736(05)70282-8. [DOI] [PubMed] [Google Scholar]
- 55.Finch R G, Pritchard D I, Bycroft B W, Williams P, Stewart G S A B. J Antimicrob Chemother. 1998;42:569–571. doi: 10.1093/jac/42.5.569. [DOI] [PubMed] [Google Scholar]
- 56.Holden M, Swift S, Williams P. Trends Microbiol. 2000;8:101–103. doi: 10.1016/s0966-842x(00)01718-2. [DOI] [PubMed] [Google Scholar]