

Abstract
We present here a method for in vivo transposon mutagenesis of a methanogenic archaeon, Methanosarcina acetivorans C2A, which because of its independence from host-specific factors may have broad application among many microorganisms. Because there are no known Methanosarcina transposons we modified the mariner transposable element Himar1, originally found in the insect Hematobia irritans, to allow its use in this organism. This element was chosen because, like other mariner elements, its transposition is independent of host factors, requiring only its cognate transposase. Modified mini-Himar1 elements were constructed that carry selectable markers that are functional in Methanosarcina species and that express the Himar1 transposase from known Methanosarcina promoters. These mini-mariner elements transpose at high frequency in M. acetivorans to random sites in the genome. The presence of an Escherichia coli selectable marker and plasmid origin of replication within the mini-mariner elements allows facile cloning of these transposon insertions to identify the mutated gene. In preliminary experiments, we have isolated numerous mini-mariner-induced M. acetivorans mutants, including ones with insertions that confer resistance to toxic analogs and in genes that encode proteins involved in heat shock, nitrogen fixation, and cell-wall structures.
The strictly anaerobic, methane-producing Archaea (methanoarchaea) are responsible for virtually all biogenic methane production, which has been estimated at ca. 5 × 1014 g of methane per year (1). This methanogenic process is critically important as a key step in the global carbon cycle, and because it results in the production of a significant greenhouse gas. It is also at the center of alternative fuel strategies and plays an important role in both agriculture and the waste treatment industry.
Although the biochemistry of methanogenesis has been studied in considerable detail (reviewed in refs. 2 and 3), our overall knowledge of this intriguing group of organisms remains rudimentary at best. A major factor contributing to our ignorance of methanoarchaeal biology is the lack of methods for genetic analysis of these organisms. This problem has been alleviated somewhat in recent years by the development of genetic tools for use in members of the two distantly related genera, Methanococcus and Methanosarcina. These tools include selectable markers for genetic crosses (4–7), methods for high efficiency transformation (8, 9), plasmid shuttle vectors (8, 10), and methods for gene replacement (4, 11, 12) (the latter only in Methanococcus species). Despite these advances there continues to be a need for development of new tools for genetic analysis of methanoarchaea. In particular, we have been interested in developing a method for random, selectable mutagenesis (such as transposon mutagenesis) of the Methanosarcina species.
Most genetic studies of methanoarchaea published to date have involved directed mutagenesis of previously identified genes using gene replacement methods (4, 11–13). Although such studies are invaluable for examining the roles of specific genes in specific functions, they are of limited use in identifying new roles for these genes, because one must have a specific trait in mind when testing the phenotypic consequences of mutations in a given gene. For the same reason, directed mutagenesis is also of limited use in identifying the roles of genes of unknown function. Studies of this nature are much more easily accomplished by using a random mutagenesis technique. In these studies, the entire genome is mutagenized in a random fashion, and mutants defective in a specific trait are isolated based on screening or selection for mutants displaying a particular phenotype. Studies using random mutagenesis to identify genes involved in specific functions in methanoarchaea are extremely scarce. We are aware of only two such studies, both in Methanococcus maripaludis. In one study, acetate auxotrophs were identified by screening mutants created by random insertion of nonreplicating plasmids (14). In the other, a cloned DNA segment was mutagenized in Escherichia coli with a modified transposable element. The transposon-induced mutants then were recombined back onto the M. maripaludis chromosome to identify genes involved in nitrogen fixation (12). This “shuttle mutagenesis” technique demonstrates the utility of transposons in random mutagenesis studies; however, it is limited by the need to clone the target DNA into a suitable host, such as E. coli, before mutagenesis. Thus, the method is subject to cloning biases. Furthermore, multiple lengthy steps are required before the stage where transposon insertions can be screened for a phenotype. A method that would allow in vivo transposon mutagenesis in the organism under study would circumvent both difficulties. Unfortunately, no method for in vivo transposon mutagenesis has been reported for any methanoarchaeon to date.
The ability to tag mutations with known sequence elements or with selectable antibiotic resistance genes has made transposon mutagenesis one of the most powerful techniques in genetics (see for example refs. 15 and 16). Genetic methods involving transposons are widespread in both bacteria and eukaryotes. However, developing transposon systems for in vivo mutagenesis of poorly studied organisms is not a simple task. One of the major difficulties in developing a new system for transposon mutagenesis stems from the observation that most transposons require accessory host factors for efficient transposition. Therefore, most transposons do not transpose well (or at all) in distantly related species that lack proteins with sufficient similarity to the essential host factors (15, 16). A way around this problem is to use a native transposon; however, this creates additional problems. Elements derived from the organism under study often preferentially insert into resident copies of the transposon via homologous recombination, rather than into novel sites via a transposition mechanism (17). An additional problem associated with the use of native transposons is the trait of transposition immunity, in which a resident element can suppress transposition of additional incoming elements (15). These findings suggest that the ideal candidate for use in developing a new transposon tool would be an element from an organism related to the organism under study. Unfortunately, a candidate transposon that fits this criterion is rarely available, particularly for use in less-well characterized systems such as the methanoarchaea.
Recently, a method for development of new transposon tools based on the transposons of the mariner family has emerged that circumvents the problems associated with the use of native elements. The mariner family of transposons, first identified in Drosophila mauritiana, is known to be widespread in nature (18, 19). A key feature that separates these elements from many other transposons is their independence from species-specific host factors for transposition (20, 21). Thus, in vitro transposition reactions containing only target DNA, transposon DNA, and purified mariner transposase catalyze cut-and-paste transposition in a very efficient manner. This feature has allowed mariner elements to be used for mutagenesis of a wide variety of non-native organisms, including other insects (22), zebrafish (23), protozoa (24), chickens (25), and human cells (in culture) (26). Even more impressive, versions of the element Himar1, originally isolated from the hornfly, Hematobia irritans, that express the transposase gene from appropriate bacterial promoters function in E. coli and Mycobacterium smegmatis (27). Encouraged by the fact that Himar1 derivatives could transpose in organisms separated by such vast evolutionary distances, we attempted to construct derivatives that might be functional in the methanoarchaeon, Methanosarcina acetivorans. In this study, we report that such derivatives are indeed functional in vivo in M. acetivorans and that they can be used in the genetic analysis of this poorly understood organism.
Materials and Methods
Bacterial Strains, Media, and Growth Conditions.
Standard conditions were used for growth of E. coli strains (28). DH5α (Life Technologies, Gaithersburg, MD) and DH5α/λpir (29) were from S. Maloy (University of Illinois, Urbana). The former was used as the host for most plasmids; the latter was used as the host for pir-dependent replicons. M. acetivorans C2A (= DSM 2834) and Methanosarcina barkeri Fusaro (= DSM 804) were from laboratory stocks. Methanosarcina strains were grown in single cell morphology (30) at 35°C in high salt broth medium containing 125 mM methanol plus 40 mM sodium acetate (high salt-MA medium) (31). Growth of M. acetivorans on media solidified by addition of 1.5% agar was as described in ref. 30 with the modifications described in ref. 6. All plating manipulations were carried out under strictly anaerobic conditions in an anaerobic glove box. Solid media plates were incubated in an intrachamber anaerobic incubator as described (32). Puromycin was added to media as indicated at 2 μg/ml for selection of Methanosarcina strains carrying the pac gene. Fluoroacetic acid (FAA) or bromoethanesulfonic acid (BES) were added from sterile, anaerobic stock solutions for selection of mutants resistant to each compound. Final concentrations used were 50 mM FAA in high salt medium with 125 mM methanol and 10 mM sodium pyruvate, or 1.6 mM BES in high salt-MA medium.
DNA Methods.
Standard methods were used throughout for isolation and manipulation of plasmid DNA from E. coli (33). Genomic DNA isolation from M. acetivorans and DNA hybridizations were performed as described (6, 31). Probes used for hybridization experiments were labeled with α-32P-dATP using the Prime-a-Gene kit (Promega) according to specifications. DNA sequences were determined from double-stranded templates by automated dye terminator sequencing. DNA sequencing was performed at the W.M. Keck Center for Comparative and Functional Genomics, University of Illinois. Primers used for determination of transposon insertion junctions were: pJK5forI, 5′-GTATATTACGAATAGGGCG-3′, and pJK5revI, 5′-AGCTGCTGGTGAAAGAGAC-3′.
Transformations.
E. coli strains were transformed by electroporation using an E. coli Gene Pulser (Bio-Rad) as recommended. Liposome-mediated transformation was used for Methanosarcina species as described in ref. 8 with the modifications described in ref. 6.
Plasmid Constructions.
Plasmid pJK5 carries a restriction endonuclease cassette (the pac-ori-aph cassette) that includes the aph gene (which encodes kanamycin resistance in E. coli), the origin of replication from plasmid R6K (which is functional in enteric bacteria that provide the pir gene in trans; ref. 29), and the pac gene under the control of the Methanococcus voltae mcr promoter (which encodes resistance to puromycin in Methanosarcina species; ref. 8). Plasmid pJK5 was constructed in two steps. First, pJK4 was constructed by ligation of the 441-bp EcoRI to BamHI fragment from pGP704 (29) with the 1,240-bp PstI aph cassette of pUC4K (Amersham Pharmacia) after treatment of both fragments with DNA polymerase I Klenow fragment and deoxynucleoside triphosphates (dNTPs). Plasmid pJK5 was constructed by ligation of RcaI-cut pJK4 into the NcoI site of pJK3 (8).
Plasmids pWM366 and pWM367, which carry the mini-mariner elements mini-MAR366 and mini-MAR367, respectively, were constructed by ligation of the 2,969-bp KpnI to XbaI pac-ori-aph cassette of pJK5, made blunt by treatment with T4 DNA polymerase and dNTPs, into SmaI-cut pMini-mariner (34). The two plasmids, and their respective mini-mariner elements, differ only with respect to the orientation of the cassette within the mariner inverted repeats.
Plasmids pWM368, pWM369, and pWM370 were constructed to allow expression of the Himar1 transposase (tnp) from promoters of varying strength in M. acetivorans. Each is a derivative of pMarNde18 (20) in which the natural Himar1 transposase promoter is replaced by a M. barkeri Fusaro promoter. In each plasmid, the tnp ATG start codon is fused directly to the ATG start codon of the Methanosarcina gene in question. Thus, both transcription and translation of the tnp gene are under the control of Methanosarcina regulatory elements. The promoter region of each gene was amplified from M. barkeri Fusaro genomic DNA by using Taq DNA polymerase and the primers indicated. The mcrB promoter (35) was amplified by using the primers 5′-CGCGCGTACGCGTGCATGCTTAAAAAAATACATAAATTCAATTATCGGAG-3′ and 5′-CGCGCGTACGCGTCATATGAATTTCCTCCTTAATTTATTAAAATCATTTTGG-3′; the orf2 promoter (31) was amplified with the primers 5′-CGCGCGGCATGCGGATCACAGTCCTTTGAATG-3′ and 5′-CGCGCGCATATGCACCGACTCCTTGTTTAATGT3′, and the serC promoter (31) was amplified with the primers 5′-CGCGCGCATATGATCTTTTCCTTTTTTGGTACTG-3′ and 5′-CGCGCGGCATGCGATC TCAACCACCTTTTTTCC-3′. Each PCR product was digested with SphI and NdeI (restriction sites added to the PCR primers are shown in bold) and cloned into the same sites in pMarNdeI. Plasmids pWM368, pWM369, and pWM370 contain the mcrB, serC, and orf2 promoters, respectively.
A series of plasmids were constructed as suicide vectors for delivery of the mini-MAR elements and the modified tnp genes into M. acetivorans. To construct these plasmids the 3.2-kbp XbaI to SpeI fragment of pWM366 carrying the mini-MAR366 element was ligated into the XbaI site immediately upstream of the tnp gene in the plasmids pMarNdeI, pWM368, pWM369, and pWM370 to create pWM379, pWM381, pWM383, and pWM385, respectively. Plasmid pWM382 is identical to pWM381, but with mini-MAR366 in the opposite orientation. Plasmids pWM397 and pWM398 are identical to pWM381 and pWM382, but carry the mini-MAR367 element, isolated as a 3.2-kbp XbaI to SpeI fragment from pWM367, in place of mini-MAR366. Finally, four additional vectors, pJK57, pJK58, pJK59, and pJK60, are otherwise identical to plasmids pWM381, pWM382, pWM397, and pWM398, but carry two mutations of the tnp gene that result in higher transposition frequencies relative to wild-type transposase. These plasmids were constructed by replacement of the 810-bp EcoRV to PstI fragments within the tnp gene of pWM381, pWM382, pWM397, and pWM398 with the same fragment from plasmid pBADC9 (36).
Nucleotide sequences of all plasmids described are available on request.
Results
Construction of a Himar1 mariner Transposon System for Use in M. acetivorans.
To allow its use in M. acetivorans, we modified the Himar1 element as described in Materials and Methods to provide an appropriate selectable marker and to allow expression of the Himar1 transposase gene. The structures of the modified mini-mariner elements, mini-MAR366 and mini-MAR367, are shown in Fig. 1A. There are several important features in each transposon that are critical for their use in M. acetivorans. The pac cassette allows isolation of strains carrying transposon insertions into the M. acetivorans genome by selection for puromycin resistance (PurR). The aph gene and origin of replication from the plasmid R6K allow facile cloning of these transposon insertions in E. coli (see below).
Figure 1.
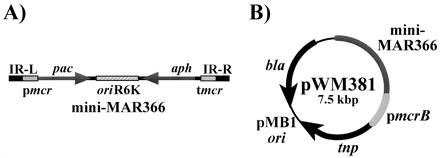
(A) The physical structure of the modified mini-mariner transposon mini-MAR366. The left and right mariner inverted repeats (IR-L and IR-R) flank the pac gene, which encodes resistance to puromycin in Methanosarcina, the E. coli plasmid R6K origin of replication, and the aph gene, which encodes resistance to kanamycin in E. coli. Transcription of the pac gene in Methanosarcina is from the M. voltae methyl reductase operon promoter, pmcr. The transcription terminator tmcr is located immediately upstream of the aph gene. Mini-MAR367 (not shown) is identical to mini-MAR366, except that the region between the inverted repeats is in the opposite orientation. (B) The physical structure of the mini-mariner delivery plasmid pWM381. This plasmid replicates in E. coli by virtue of the high-copy plasmid pMB1 replicon, but cannot replicate in Methanosarcina. The gene encoding the mariner transposase, tnp, is expressed from the strongly transcribed pmcrB promoter of M. barkeri. Plasmids pWM379, pWM383, and pWM385 (not shown) are identical to pWM381, except that the pmcrB promoter is replaced by one of three alternate promoters, as described in Materials and Methods. Additional elements described in the text vary with respect to the orientation of the mini-MAR element; some carry mini-MAR367 in place of mini-MAR366.
To allow expression of the Himar1 tnp gene in M. acetivorans we constructed a series of plasmids that place its expression under the control of promoters of varying strengths from the closely related organism M. barkeri Fusaro. The promoters we used were the highly expressed mcrB promoter (35), the moderately expressed serC promoter and the poorly expressed orf2 promoter (31). A plasmid bearing the native Himar1 tnp promoter also was included (20).
Lastly, the mini-mariner elements and modified tnp genes were combined to create a series of suicide vectors for delivery of the transposons into M. acetivorans (Fig. 1B). These delivery plasmids carry the pMB1 replicon and, thus, can be isolated in large quantities from E. coli; however, the plasmids do not replicate in M. acetivorans, nor do they carry any known sequences homologous to the chromosome of M. acetivorans. Therefore, PurR clones obtained after transformation of M. acetivorans with a delivery plasmid should represent transposition events of the mini-mariner element into the M. acetivorans chromosome. We chose to construct our transposon system so that the transposase gene would not be included within the transposable element itself, but would instead be provided in cis immediately adjacent to a mini-mariner transposon. Transposon insertions obtained with the system should not contain the tnp gene and, thus, will be incapable of further transposase-mediated events. Therefore, these insertions should be stable even in the absence of continued antibiotic selection.
Transposition of mini-MAR Elements in M. acetivorans.
Initially, the mini-mariner transposons were introduced into M. acetivorans by transformation with a series of the suicide delivery plasmids that differ only in the promoter used for expression of the tnp gene. In these experiments, hundreds of PurR colonies were obtained after transformation with delivery plasmids that express tnp from the highly expressed mcrB promoter (Table 1). Approximately 4-fold fewer PurR colonies were obtained with plasmids that express tnp from either the orf2 or the serC promoter. PurR colonies were never observed when tnp was expressed from its native Himar1 promoter (data not shown).
Table 1.
Transformation of M. acetivorans with various mini-MAR delivery plasmids
| Plasmid | Element (orientation) | PurR colonies/μg DNA* |
|---|---|---|
| pWM381 | mini-MAR366 (+) | 58 ± 43 |
| pWM382 | mini-MAR366 (−) | 84 ± 48 |
| pWM397 | mini-MAR367 (+) | 91 ± 49 |
| pWM398 | mini-MAR367 (−) | 95 ± 50 |
| pJK57 | mini-MAR366 (+) | 278 ± 142 |
| pJK58 | mini-MAR366 (−) | 258 ± 103 |
| pJK59 | mini-MAR367 (+) | 236 ± 102 |
| pJK60 | mini-MAR367 (−) | 208 ± 159 |
Approximately 109 cells were transformed with 2 μg of each of the indicated plasmids as described in Materials and Methods. All plasmids express the tnp gene from the strongly expressed M. barkeri Fusaro mcrB promoter. Plasmids pJK57, pJK58, pJK59, and pJK60 are identical to pWM381, pWM382, pWM397, and pWM398, respectively, except that each carries a hyperactive mutant derivative of the tnp gene. Orientation of the mini-MAR element is arbitrarily defined. The + orientation has the right inverted repeat (IR-R) closer to the 5′ end of the tnp gene. The − orientation has the left inverted repeat (IR-L) element closer to the 5′ end of the tnp gene.
The numbers shown are the average and SD of at least four trials.
Because certain transposons display a preference for the relative orientation of the inverted repeats with respect to the transposase gene, we performed additional experiments with delivery vectors that varied the relative orientation of plasmid elements (Table 1). The delivery plasmids used for these experiments all expressed the tnp gene from the mcrB promoter because they showed the highest transformation frequencies in preliminary experiments. To examine the effect of the orientation of the insert within the transposable element we compared plasmids containing mini-MAR366 to mini-MAR367 (compare pWM381 with pWM397 or pWM382 with pWM398). To examine the effect of the orientation of each element with respect to the tnp gene we compared plasmids with each element inserted in opposite orientations (compare pWM381 with pWM382 or pWM397 with pWM398). No significant differences were observed, either with respect to the element used or to the orientation of each element with respect to tnp.
During the course of this study, hyperactive mutants of the Himar1 tnp gene were isolated (36). Transformation of delivery plasmids that carry these hyperactive tnp mutations resulted in ca. 3-fold higher numbers of PurR transformants than otherwise identical plasmids (compare plasmids pWM381, pWM382, pWM397, and pWM398 with pJK57, pJK58, pJK59, and pJK60). These increases are similar to those seen in in vitro transposition reactions using the mutant Tnp protein (36). In previous studies, we have shown the transformation method used here gives an average of ca. 107 transformants/μg DNA using replicating plasmids (8). Therefore, the transposition frequency in cells that receive these improved delivery vectors is ca. 2.5 × 10-5 (transposition frequency = number of colonies obtained with the delivery vector/number of colonies transformed).
To verify that the PurR colonies obtained after transformation with the delivery plasmids had transposon insertions into the M. acetivorans chromosome we examined genomic DNA from 20 PurR colonies by DNA hybridization. As shown in Fig. 2, hybridization signals from each clone were seen when a probe homologous to the mini-MAR element was used; however, no signal was observed when a probe homologous to the delivery vector backbone was used. This indicates that the delivery vector was lost whereas the transposon was retained. Furthermore, the hybridizing bands seen with the mini-MAR probe were of various sizes indicating that the mini-MAR elements were inserted at different sites within the genome.
Figure 2.
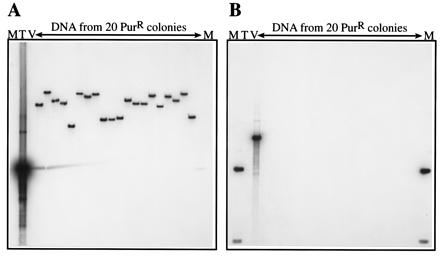
Verification of mini-MAR transposition by DNA hybridization. Genomic DNA from 20 PurR colonies obtained by transformation of M. acetivorans with the delivery plasmid pWM381 was digested with EcoRI (which does not cut within the transposon), electrophoresed, and blotted onto nylon membranes as described in Materials and Methods. The membranes then were examined by hybridization with two probes, one specific for the transposon only (A), the other specific for the delivery vector backbone only (B). Each of the 20 transposon insertions was at a different site within the M. acetivorans genome as shown by the different-sized bands in A. None of the 20 insertions hybridized to the vector probe, indicating that the delivery plasmid backbone was lost in all cases (B). The plasmids that were used as probes also were included in the blot as positive controls for hybridization and as molecular weight markers. pBluescript SK+ (2.9 kbp), which hybridizes only to the vector backbone, was used as a vector-specific probe (V), and pJK4 (1.6 kbp), which hybridizes only to the mini-MAR insert, was used as a transposon-specific probe (T). The 1-kbp ladder (Life Technologies) is shown by M, the hybridizing band in B is 1.6 kbp.
Cloning and DNA Sequence Analysis of mini-MAR Insertions.
The mini-MAR elements were designed to allow facile cloning of transposon insertion sites as shown in Fig. 3. Cloning of transposon insertions is accomplished by cutting genomic DNA from a transposon-induced mutant with an enzyme (such as EcoRI) that does not cut within the transposon. Subsequently, the digested DNA is treated with DNA ligase and introduced into pir+ E. coli, where it replicates as a plasmid. The junction sequences of the transposon insertions in these plasmids can easily be determined by using outward-directed, transposon-specific primers.
Figure 3.
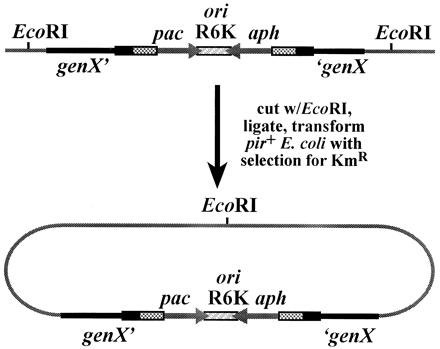
Cloning of mini-MAR insertions. The chromosomal region (gray line) surrounding a mini-MAR insertion into genX (black line) can be cloned directly from genomic DNA by cutting with a restriction endonuclease that does not cut within the element (EcoRI for example), followed by ligation and transformation into a pir+ strain of E. coli where the circularized DNA is capable of replicating as a kanamycin-resistance plasmid.
We cloned and sequenced the mini-MAR insertion junctions with genomic DNA from each of the 20 clones used in the hybridization experiments described above (Table 2). Analysis of these DNA sequences indicates that each of the 20 insertions occurred at a different site within the M. acetivorans chromosome. In no case were sequences related to the delivery vector obtained, indicating that all insertions arose by transposition rather than by some form of illegitimate recombination. Further, each insertion examined occurred at a TA dinucleotide with duplication of two bases at each transposon junction. These features are seen in all mariner transposition sites identified to date (34). Although the majority (12/20) of the insertions were in genes with no previously identified homologs, eight of the insertions were in genes that were very similar to known genes (Table 2). In six of eight of these cases, the strongest matches were to archaeal homologs. In two cases, the homologous gene has been identified in a closely related Methanosarcina species. In sum, these data clearly indicate that the mini-mariner elements are transposing in vivo in M. acetivorans C2A.
Table 2.
DNA sequence analysis of mini-MAR insertions
| Clone no. | Closest homolog | Accession no. | blast score |
|---|---|---|---|
| 6 | Methanococcus jannaschii (MJ1507) | Q58902 | 4e-24 |
| hypothetical orf | |||
| 7 | Pyrococcus horikoshii (PH0606) | E71104 | 0.005 |
| Cdc47 homolog* | |||
| 10 | Methanosarcina mazei | P35515 | 3e-47 |
| DnaJ | |||
| 13 | Methanobacterium thermoautotrophicum | O27585 | 2e-11 |
| lysS* | |||
| 14 | Anabaena variabilis | U51863 | 5e-13 |
| VnfN | |||
| 17 | E. coli | P27848 | 7e-04 |
| hypothetical orf yigL | |||
| 18 | M. mazei | Y12024 | le-15 |
| orf46 S-layer protein | |||
| 19 | Acetobacter plasmid pAH4 | JC2322 | 0.003 |
| hypothetical orf |
The 20 mini-MAR insertions analyzed by DNA hybridization were cloned, and the junction sequences were determined as described in Fig. 3. The DNA sequence adjacent to the mini-MAR element (typically ca. 300–400 bp) were used to search the National Center for Biotechnology Information nonredundant protein database by using the BlastX algorithm on 4/19/2000. Eight of the 20 clones returned homologous sequences with P values < 0.005. The best score for each of these is listed above along with the associated SwissProt accession numbers and BlastX P values.
Disruption of these genes may reasonably be expected to be lethal; however, in these cases the insertion is adjacent to, but not within, the homologous gene.
Use of Mini-MAR Elements to Isolate M. acetivorans Mutants Resistant to BES and FAA.
The coenzyme M (CoM) analog BES is a potent inhibitor of the enzyme methyl-CoM reductase that catalyzes the final step in methane production. In earlier studies, M. voltae strains incapable of either BES or CoM uptake were isolated as BESR mutants (37). However, the mutated gene(s) in these strains were never identified. To demonstrate the utility of the mini-MAR transposons system in genetic analysis of M. acetivorans, we isolated transposon-induced BESR mutants and identified the disrupted genes by cloning and sequencing as described above. To do this, we screened several thousand mini-MAR-induced mutants selected as PurR colonies by replica plating onto solid medium containing BES. We identified two BESR mutants that grow well in medium containing four times the minimal inhibitory concentration (MIC) of BES for wild-type M. acetivorans (the MIC is ca. 0.4 mM). Sequence analysis of the cloned insertions indicates these mutants have mini-MAR-366 insertions into an apparent operon encoding proteins with strong homology to a family of ABC transporters that includes known sulfate permeases (data not shown). We believe that this putative transporter is probably involved in the uptake of sulfur containing molecules such as sulfonates (CoM and BES) and/or sulfate. In a similar way, we identified two mini-MAR-induced mutations that confer resistance to the toxic acetate analog FAA. The mutants carrying these insertions grow well in medium containing ca. five times the wild-type MIC for FAA. FAAR mutants have been isolated in numerous microorganisms. Commonly these mutations lie in the genes responsible for entry of acetate into central metabolism, namely pta (which encodes the enzyme phosphotransacetylase) and ack (which encodes the enzyme acetate kinase) (38). Sequence analysis of the mini-MAR-induced FAAR M. acetivorans mutants indicates that both have insertions into the pta-ack operon of this organism (data not shown). Detailed studies of these mutants will be reported elsewhere.
Discussion
Development of genetic methods applicable to diverse organisms has not kept pace with the rapid development of molecular biological methods. Thus, we now are faced with numerous situations where microbial genomes have been completely sequenced for organisms in which few or no genetic methods exist. Examination of these genome sequences has led to predictions concerning the roles of various genes in the metabolism of the organisms where they are found. However, it is important to remember that these are only predictions. Experimental data on the function of these genes (i.e., genetics) will be absolutely required to test these predictions. Therefore, it is essential that genetic methods be developed for these diverse organisms.
We have been attempting to develop methods for genetic analysis for members of the archaeal genus Methanosarcina. A long-time goal of this research has been the development of selectable transposons for use in mutagenesis of this organism. In this study, we showed that the insect mariner-family transposon Himar1 could be modified to function in vivo in M. acetivorans. The elements we constructed transpose at relatively high frequencies, comparable to those of bacterial transposons (39, 40). The transposons insert at essentially random loci in the genome, with the only target specificity being the absolute requirement for a TA dinucleotide at the insertion site that is seen for all mariner transposition events (34). Further, we have demonstrated the utility of these mariner elements in genetic analysis of M. acetivorans by isolating loss-of-function mutations in a known catabolic gene and in a novel ABC family transporter. In addition to the mutations presented here, we have isolated numerous strains with mini-MAR-induced mutations that display clear phenotypes. For example, we have isolated a mutant resistant to toxic base analogs with an insertion into the hpt gene, encoding hypoxanthine phosphoribosyl transferase and several insertions that interfere with the cells ability to use methanol as a methanogenic substrate (data not shown).
The study presented here demonstrated in vivo transposition in just a single host species, M. acetivorans C2A; however, we believe the elements we developed may find widespread use in other archaeal species as well. In principal, the mini-MAR elements should be functional in any organism in which the promoters and selectable markers we used are functional. It is known that archaeal promoters can work in widely divergent species, and the pac gene we use here is functional in distantly related Methanococcus species (41). (For comparison, the evolutionary distance between Methanosarcina and Methanococcus is roughly the same as between humans and slime molds.) Therefore, we believe that these mini-MAR elements and delivery vectors should be useful in other archaeal species for which high-efficiency transformation protocols are available. This belief has recently been confirmed by demonstration that they are functional in vivo in M. maripaludis (J. A. Leigh and W. B. Whitman, personal communications).
Although originally isolated in a eukaryote, the Himar1 transposon has been shown to function in vivo in bacteria (27). Our finding that it can also function in Archaea extends the Himar1 host range to organisms in each of the three domains of life. This observation has profound implications with respect to horizontal gene transfer and evolution. Transposons are well known for their ability to catalyze various genetic rearrangements, including mobilization of genes for transfer to new loci. The finding that certain transposons are functional across such vast evolutionary distances suggests a mechanism for gene transfer across those same distances. In this regard, it is important to note that in our study we failed to observe transposition when the tnp gene was expressed from its native promoter. Thus, such horizontal transfer may be reduced considerably by the inability of promoters to function in diverse hosts.
Acknowledgments
We thank John Leigh (University of Washington) and William Whitman (University of Georgia) for sharing unpublished data. This work was supported by Grant MCB-987459 from the National Science Foundation and a Searle Scholars Award to W.W.M. and by Grant AI33586 from the National Institutes of Health to D.J.L. and H.M.R.
Abbreviations
- FAA
fluoroacetic acid
- BES
bromoethanesulfonic acid
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160272597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160272597
References
- 1.Rogers J E, Whitman W B, editors. Microbial Production and Consumption of Greenhouse Gases: Methane, Nitrogen Oxides, and Halomethanes. Washington, DC: Am. Soc. Microbiol.; 1991. [Google Scholar]
- 2.Ferry J G, editor. Methanogenesis: Ecology, Physiology, Biochemistry, and Genetics. New York: Chapman and Hall; 1993. [Google Scholar]
- 3.Deppenmeier U, Lienard T, Gottschalk G. FEBS Lett. 1999;457:291–297. doi: 10.1016/s0014-5793(99)01026-1. [DOI] [PubMed] [Google Scholar]
- 4.Gernhardt P, Possot O, Foglino M, Sibold L, Klein A. Mol Gen Genet. 1990;221:273–279. doi: 10.1007/BF00261731. [DOI] [PubMed] [Google Scholar]
- 5.Argyle J L, Tumbula D L, Leigh J A. Appl Environ Microbiol. 1996;62:4233–4237. doi: 10.1128/aem.62.11.4233-4237.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boccazzi P, Zhang J K, Metcalf W W. J Bacteriol. 2000;182:2611–2618. doi: 10.1128/jb.182.9.2611-2618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfeiffer M, Bestgen H, Burger A, Klein A. Arch Microbiol. 1998;170:418–426. doi: 10.1007/s002030050662. [DOI] [PubMed] [Google Scholar]
- 8.Metcalf W W, Zhang J K, Apolinario E, Sowers K R, Wolfe R S. Proc Natl Acad Sci USA. 1997;94:2626–2631. doi: 10.1073/pnas.94.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tumbula D L, Makula R A, Whitman W B. FEMS Microbiol Lett. 1994;121:309–314. [Google Scholar]
- 10.Tumbula D L, Bowen T L, Whitman W B. J Bacteriol. 1997;179:2976–2986. doi: 10.1128/jb.179.9.2976-2986.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berghofer Y, Klein A. Appl Environ Microbiol. 1995;61:1770–1775. doi: 10.1128/aem.61.5.1770-1775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blank C E, Kessler P S, Leigh J A. J Bacteriol. 1995;177:5773–5777. doi: 10.1128/jb.177.20.5773-5777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarrell K F, Bayley D P, Florian V, Klein A. Mol Microbiol. 1996;20:657–666. doi: 10.1046/j.1365-2958.1996.5371058.x. [DOI] [PubMed] [Google Scholar]
- 14.Kim W, Whitman W B. Genetics. 1999;152:1429–1437. doi: 10.1093/genetics/152.4.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg D E, Berg C M. Biotechnology. 1983;1:417–435. [Google Scholar]
- 16.Kleckner N, Bender J, Gottesman S. Methods Enzymol. 1991;204:139–180. doi: 10.1016/0076-6879(91)04009-d. [DOI] [PubMed] [Google Scholar]
- 17.Dyall-Smith M L, Doolittle W F. Can J Microbiol. 1994;40:922–929. doi: 10.1139/m94-148. [DOI] [PubMed] [Google Scholar]
- 18.Plasterk R H. Curr Top Microbiol Immunol. 1996;204:125–143. doi: 10.1007/978-3-642-79795-8_6. [DOI] [PubMed] [Google Scholar]
- 19.Robertson H M. J Insect Physiol. 1995;41:99–105. [Google Scholar]
- 20.Lampe D J, Churchill M E, Robertson H M. EMBO J. 1996;15:5470–5479. [PMC free article] [PubMed] [Google Scholar]
- 21.Tosi L R, Beverley S M. Nucleic Acids Res. 2000;28:784–790. doi: 10.1093/nar/28.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coates C J, Jasinskiene N, Miyashiro L, James A A. Proc Natl Acad Sci USA. 1998;95:3748–3751. doi: 10.1073/pnas.95.7.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fadool J M, Hartl D L, Dowling J E. Proc Natl Acad Sci USA. 1998;95:5182–5186. doi: 10.1073/pnas.95.9.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gueiros-Filho F J, Beverley S M. Science. 1997;276:1716–1719. doi: 10.1126/science.276.5319.1716. [DOI] [PubMed] [Google Scholar]
- 25.Sherman A, Dawson A, Mather C, Gilhooley H, Li Y, Mitchell R, Finnegan D, Sang H. Nat Biotechnol. 1998;16:1050–1053. doi: 10.1038/3497. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Sankar U, Lampe D J, Robertson H M, Graham F L. Nucleic Acids Res. 1998;26:3687–3693. doi: 10.1093/nar/26.16.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubin E J, Akerley B J, Novik V N, Lampe D J, Husson R N, Mekalanos J J. Proc Natl Acad Sci USA. 1999;96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wanner B L. J Mol Biol. 1986;191:39–58. doi: 10.1016/0022-2836(86)90421-3. [DOI] [PubMed] [Google Scholar]
- 29.Miller V L, Mekalanos J J. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sowers K R, Boone J, Gunsalus R P. Appl Environ Microbiol. 1993;59:3832–3839. doi: 10.1128/aem.59.11.3832-3839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalf W W, Zhang J K, Shi X, Wolfe R S. J Bacteriol. 1996;178:5797–5802. doi: 10.1128/jb.178.19.5797-5802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metcalf W W, Zhang J K, Wolfe R S. Appl Environ Microbiol. 1998;64:768–770. doi: 10.1128/aem.64.2.768-770.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ausebel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1992. [Google Scholar]
- 34.Lampe D J, Grant T E, Robertson H M. Genetics. 1998;149:179–187. doi: 10.1093/genetics/149.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allmansberger R, Bokranz M, Krockel L, Schallenberg J, Klein A. Can J Microbiol. 1989;35:52–57. doi: 10.1139/m89-008. [DOI] [PubMed] [Google Scholar]
- 36.Lampe D J, Akerley B J, Rubin E J, Mekalanos J J, Robertson H M. Proc Natl Acad Sci USA. 1999;96:11428–11433. doi: 10.1073/pnas.96.20.11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoro N, Konisky J. J Bacteriol. 1987;169:660–665. doi: 10.1128/jb.169.2.660-665.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown T D, Jones-Mortimer M C, Kornberg H L. J Gen Microbiol. 1977;102:327–336. doi: 10.1099/00221287-102-2-327. [DOI] [PubMed] [Google Scholar]
- 39.Reznikoff W S. Annu Rev Microbiol. 1993;47:945–963. doi: 10.1146/annurev.mi.47.100193.004501. [DOI] [PubMed] [Google Scholar]
- 40.Huisman O, Kleckner N. Genetics. 1987;116:185–189. doi: 10.1093/genetics/116.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen-Kupiec R, Marx C J, Leigh J A. J Bacteriol. 1999;181:256–261. doi: 10.1128/jb.181.1.256-261.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]