

Abstract
Ethionamide (ETA) is an important component of second-line therapy for the treatment of multidrug-resistant tuberculosis. Synthesis of radiolabeled ETA and an examination of drug metabolites formed by whole cells of Mycobacterium tuberculosis (MTb) have allowed us to demonstrate that ETA is activated by S-oxidation before interacting with its cellular target. ETA is metabolized by MTb to a 4-pyridylmethanol product remarkably similar in structure to that formed by the activation of isoniazid by the catalase-peroxidase KatG. We have demonstrated that overproduction of Rv3855 (EtaR), a putative regulatory protein from MTb, confers ETA resistance whereas overproduction of an adjacent, clustered monooxygenase (Rv3854c, EtaA) confers ETA hypersensitivity. Production of EtaA appears to be negatively regulated by EtaR and correlates directly with [14C]ETA metabolism, suggesting that EtaA is the activating enzyme responsible for thioamide oxidation and subsequent toxicity. Coding sequence mutations in EtaA were found in 11 of 11 multidrug-resistant MTb patient isolates from Cape Town, South Africa. These isolates showed broad cross-resistance to thiocarbonyl containing drugs including ETA, thiacetazone, and thiocarlide.
In 1998 Mycobacterium tuberculosis (MTb) infected 7.25 million people and resulted in 2.9 million fatalities (1). Underlying these statistics is an emerging epidemic of multidrug-resistant tuberculosis (MDRTB) that severely undermines control efforts and is transmitted indiscriminately across national borders (2, 3). As many as 50 million people worldwide presently are infected with MDRTB, defined as resistance to both isoniazid (INH) and rifampicin (4). Resistance to any of the front-line drugs such as INH and rifampicin generally bodes poorly for the patient who then is committed to a regimen of 4–5 less-active “second-line” therapies. One of the most efficacious of these second-line drugs that is widely used in the treatment of MDRTB is the thioamide ethionamide (ETA) (1) (5).
Current tuberculosis therapies include a large number of “prodrugs” that must be metabolically activated to manifest their toxicity upon specific cellular targets (6). The best-characterized example of this is the activation of INH by the catalase-peroxidase KatG that precedes inactivation of enzymes involved in mycolic acid biosynthesis (7). Although this process has been intensively studied, the actual identity of the active form of INH remains unclear (8). There appear to be competing oxidative reactions that give rise to a variety of metabolites, suggesting a highly reactive intermediate. One product that has been identified both via x-ray crystallography within the active site of InhA (one of the proposed activated INH target enzymes) (9, 10) and via direct formation under appropriate conditions in solution is the acylpyridine adduct of NAD+ (11). Despite the apparent in vitro complexity, activation of INH by whole cells of MTb gives primarily a single major product, 4-pyridylmethanol, which is in effect a surrogate for the productive activation of INH mediated by KatG (12, 13). INH metabolism to 4-pyridylmethanol only occurs in drug-susceptible organisms whereas drug-resistant organisms no longer produce this metabolite (14). The majority of clinically observed INH resistance is associated with the loss of this activating ability by the bacillus (15).
Like the front-line INH, ETA is specific for mycobacteria and is thought to exert a toxic effect on mycolic acid constituents of the cell wall of the bacillus (10, 16, 17). Although activated ETA has been shown to share a common molecular target with INH, KatG mutants resistant to INH retain their sensitivity toward ETA, suggesting that ETA activation requires an entirely different enzyme. It has long been known that in vivo ETA demonstrates more consistent antitubercular activity than one would expect from in vitro examination of its minimal inhibitory concentration (MIC), and it has been demonstrated that a major metabolite of ETA in vivo is its S-oxide (2), which shows comparable in vitro activity (18–22). A wide variety of toxic thiocarbonyl-containing compounds have been shown to be converted to the corresponding S-oxides during in vivo metabolism by mammals supporting this as an intermediate oxidation in the metabolic activation of ETA (23).
Experimental Procedures
Synthesis of 2-Ethyl-[14C]Thioisonicotinamide ([1-14C]ETA).
2-Ethylpyridine was converted to its N-oxide salt in almost quantitative yield by using 35% hydrogen peroxide in acetic acid, and the corresponding N-oxide was subjected to a nitrating mixture of sulfuric and nitric acids to form 2-ethyl-4-nitropyridine N-oxide in 60% yield (24). Reduction using iron filings, hydrochloric acid, and acetic acid (25) allowed us to isolate 2-ethyl-4-aminopyridine, which was converted to 2-ethyl-4-bromopyridine through the perbromide by using 50% aqueous hydrobromic acid and sodium nitrite (24). The resulting bromide was heated with copper [14C]cyanide in N-methylpyrrolidin-2-one to afford 2-ethyl-4-[14C]cyanopyridine (26). Copper [14C]cyanide was obtained from sodium [14C]cyanide (Amersham Pharmacia) by using copper(II) sulfate pentahydrate and sodium sulfite (27, 28). The nitrile was converted to [1-14C]ETA by hydrogen sulfide treatment, and the resulting thioamide was purified to 98% final radiochemical purity by using normal-phase HPLC with a preparative ADSORBOSPHERE silica column (5 μm, 300 × 22 mm, Alltech Associates) and an isochratic eluent of 90% chloroform, 10% methanol. Unlabeled ETA synthesized by using the same procedure cochromatographed with commercially available ETA (Sigma-Aldrich) and showed the correct analytical data.
In Vivo Metabolism of [1-14C]ETA by Whole Cells of Mycobacteria.
The indicated mycobacterial species was grown in culture to an OD650 of 1.0–1.5 and then concentrated 10-fold in middlebrook 7H9 broth media (Difco). The culture suspensions were treated with 0.01 μg/ml of [14C]ETA (55 mCi/mmol), and sequential culture aliquots were removed at the indicated times, filtered, and flash-frozen. Samples of 2 μl were analyzed by TLC on silica gel 60 plates (EM Science) developed with 95:5 ethyl acetate/ethanol. Before spotting radioactive samples on TLC plates a small amount of unlabeled ETA was spotted to circumvent silica-catalyzed air oxidation of the low concentration radioactive ETA samples.
Metabolites were identified by comparison with well-characterized synthetic standards prepared as follows: the sulfoxide (2) was prepared by hydrogen peroxide oxidation of ETA as described (29). The acid was made by reflux hydrolysis of the thioamide with 30% NaOH (Aq); 1H-NMR (CDCl3/CD4OD; 1:1); δ 1.26 t, 2.83 q, 7.63 d, 7.71 s, 8.52 d; ES-MS (MH+) 152.1 m/e. The amide (4) was made by treating the corresponding acid chloride with ammonium hydroxide; 1H-NMR (CDCl3); δ 1.34 t, 2.91 q, 7.45 d, 7.55 s, 8.65 d; ES-MS (MH+) 151.2 m/e. (2-Ethyl-pyridin-4-yl)methanol (5) was made by RedAl reduction of the acid in tetrahydrofuran; 1H-NMR (CDCl3); δ 1.28 t, 2.84 q, 4.73 s, 7.11 d, 7.19 s, 8.48 d; 13C-NMR (CDCl3); 14.12, 30.54, 63.92, 118.73, 119.64, 149.25, 150.60, 163.89; ES-MS (MH+) 138.0 m/e.
Cells from sequential culture aliquots from the metabolic conversion assays (volumes given in figure legends) were collected by filtration onto 0.22-μm GS filter disks (Millipore) under vacuum on a Hoeffer apparatus and were washed twice with 0.1 mM sodium phosphate (pH 7.5), 100 mM NaCl (500 μl). The cell-associated radioactivity was measured in 4 ml of EcoscintA scintillation solution (National Diagnostics). HPLC separation of the [14C]ETA metabolite mixture was achieved by using a reverse-phase LUNA column [5 μm, C18(2), 250 × 4.6 mm, Phenomenex, Torrence, CA] with a gradient of (0–5 min) 0% acetonitrile, 100% water; then (5–65 min) to 70% acetonitrile; then (65–80 min) to 100% acetonitrile (all solvents contained 0.1% trifluoroacetic acid). The retention time of the unknown radiolabeled major metabolite (5) using continuous radiodetection (β-RAM, INUS Systems, Tampa, FL) was used to guide cold large-scale ETA feeding experiments with up to 1 liter of log-phase MTb H37Rv, to which we fed 10 μg/ml ETA (Sigma-Aldrich). We HPLC-isolated very small quantities of unlabeled metabolite with a similar retention time to (5), by using UV254 detection. The metabolite (5) gave a mass of 137 (137.9 MH+) (mass spectrometer model API300TQMS, Perkin–Elmer/Sciex). For Mycobacterium smegmatis (MSm), macromolecule-associated radioactivity was determined by resuspending cells from microcentrifuged 900-min aliquots (400 μl) in PBS. The cells were ruptured by bead-beating (MiniBeadBeater, BioSpec Products, Bartlesville, OK, 3 × 45 sec, 0.1-mm glass beads) and extensively dialyzing the lysates with centricon 10 concentrators (Amicon) before analysis in 4 ml of EcoscintA scintillation solution.
Cloning of EtaA and EtaR.
Genomic DNA from MTb H37Rv was partially digested with Sau3AI (New England Biolabs) to give fragments of various sizes. Fragments ranging from 1 kb to 10 kb were ligated to pMV206Hyg (30) that had been previously linearized with BamHI (New England Biolabs). The ligation mixtures were electroporated into Escherichia coli DH5α (Life Technologies, Grand Island, NY) for amplification of the DNA library, which was subsequently purified and electroporated into MTb H37Rv. The resulting transformants were plated on 7H11 (Difco) agar plates that contained Hygromycin (Life Technologies, 200 μg/ml) and the indicated concentrations of ETA. Five colonies were isolated that had MICs for ETA from 2.5 to 5.0 μg/ml (the MIC for wild-type MTb is 1.0 μg/ml) (16).
EtaA and EtaR were PCR-amplified from H37Rv chromosomal DNA by using the following primers, 5′-GGGGTACCGACATTACGTTGATAGCGTGGA-3′ and 5′-ATAAGAATGCGGCCGCAACCGTCGCTAAAGCTAAACC-3′ (Rv3854c, EtaA); 5′-GGGGTACCGCACACTATCGACACGTAGTAAGC-3′ and 5′-ATAAGAATGCGGCCGCGCGGTTCTCGCCGTAAATGCT-3′ (Rv3855, EtaR), and inserted directionally into KpnI- and NotI-digested pMH29 (30).
Sequence Analysis of ETA-Resistant Clinical Isolates.
Using the aforementioned primers, EtaA was PCR-amplified from genomic DNA containing drug-resistant isolate lysates (1 ml, bead-beaten for 3 × 45 sec and aqueous-diluted 10-fold). EtaA was sequenced in its entirety by primer walking for all isolates (SEQWRIGHT, Houston, TX), and observed mutations were confirmed on both strands. For the three isolates without mutations in EtaA, EtaR and the intergenic region also were sequenced in its entirety without observing any mutations.
Results
Synthesis and in Vivo Metabolism of [14C]ETA.
We synthesized [14C]ETA from 2-ethylpyridine and sodium [14C]cyanide (see Experimental Procedures) to study the metabolism of ETA by whole cells of MTb. In the presence of live cells of MTb, ETA is converted through the S-oxide (2) to a major metabolite (5) as seen by TLC analysis of sequential time points (Fig. 1A). Metabolites corresponding to the S-oxide (2), nitrile (3), and the amide (4) were identified by cochromatography (TLC and HPLC) with standards synthesized by known methods and characterized by 1H-NMR, 13C-NMR, and MS. These metabolites were produced in small amounts by the cellular oxidation of ETA but they were the dominant products of air oxidation of ETA (compare lanes h and i in Fig. 1A).
Figure 1.
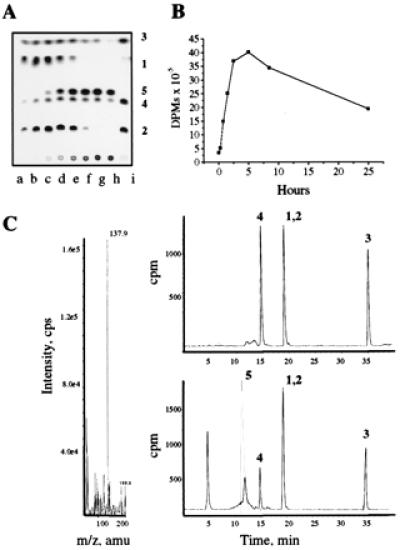
In vivo production of (2-ethyl-pyridin-4-yl)methanol (5) from ETA by whole cells of MTb. A culture of MTb strain H37Rv at OD650 of 1.0 was concentrated 10-fold in 7H9 media, and [1-14C]ETA at 0.01 μg/ml was added. (A) After incubation at 37°C metabolism of [1-14C]ETA was visualized by TLC and autoradiography. Lanes a–h correspond to sequential filtered culture samples taken at 0.2, 0.25, 0.75, 1.5, 2.5, 5.0, 8.5, and 25 h, respectively. Lane i represents media autooxidation after 25 h of incubation without bacterial cells. The metabolites observed cochromatographed with commercial and characterized synthetic samples of ETA (1), ETA S-oxide (2), ETA nitrile (3), and ETA amide (4). (B) Mycobacteria from the same sequential culture aliquots (500 μl) were collected by filtration onto 0.22-μm filter disks under vacuum, they were washed twice with PBS (500 μl), and the cell-associated radioactivity was measured. (C) The reversed-phase HPLC retention time of the unknown major metabolite (5) was used to guide cold large-scale ETA feeding experiments where we isolated unlabeled metabolite that gave a mass of 137 (137.9 MH+). We assigned this as (2-ethyl-pyridin-4-yl)methanol and confirmed the identity of (5) by cochromatography with a synthetic characterized alcohol standard. The upper HPLC continuous radiodetector spectrum corresponds to A lane i, media control and the lower spectrum; lane d, time point 1.5 h, where the UV254 trace of (2-ethyl-pyridin-4-yl)methanol is superimposed in gray.
In contrast, metabolite 5 was produced only by live cells of MTb and was not seen upon air oxidation of ETA. The thioamide S-oxide (2) was transiently produced in whole cells but was further metabolized and no longer apparent after depletion of the ETA (Fig. 1A). Cold ETA feeding experiments allowed the isolation of unlabeled metabolite 5, which displayed a molecular mass of 137 by LC-MS (Fig. 1C). We assigned this metabolite as (2-ethyl-pyridin-4-yl)methanol (5) and confirmed this by cochromatography (TLC and HPLC) with an authentic synthetic alcohol standard. The upper HPLC trace in Fig. 1C shows the continuous radio-detector output from a sample corresponding to [1-14C]ETA that has been air-oxidized in media (lane i in Fig. 1A). The lower trace shows a sample from MTb metabolism of [1-14C]ETA after 1.5 h of exposure (lane d in Fig. 1A) The UV254 trace of synthetic (2-ethyl-pyridin-4-yl)methanol is superimposed in gray. There is another unidentified more polar metabolite at the void volume of the HPLC trace that may correspond to the origin material in the TLC analysis shown in Fig. 1A. Further analysis of this material revealed that it was composed of several discrete, very polar substances. This metabolism also was associated with incorporation of ETA-derived radioactivity into whole cells (Fig. 1B).
Identification of EtaA, a Monooxygenase That Activates ETA.
To elucidate the enzymatic basis for activation of ETA to metabolite 5 by MTb we selected for ETA resistance in MTb by transformation of a 1- to 10-kb insert-containing library of MTb chromosomal DNA in pMV206Hyg (31). Five colonies were isolated that had MICs for ETA from 2.5 to 5.0 μg/ml (the MIC for wild-type MTb is 1.0 μg/ml). Upon restriction analysis the five independent plasmids were shown to contain the same genomic region on different overlapping Sau3AI fragments. This cloning also was done with genomic DNA from a strain reported to be ETA-resistant but the same genomic locus was obtained with no alterations compared with H37Rv, suggesting that the resistance was not associated with alterations to this region but simply with its overexpression. The common region to all of the resistance-conferring clones encompassed only one protein (Rv3855, EtaR) (32) that showed broad homology to many TetR family transcriptional regulators. A 76-nt intergenic region separates this putative regulator from a divergently transcribed monooxygenase (Rv3854c, EtaA). EtaA displayed significant homology to known flavin monooxygenases such as cyclohexanone monooxygenase from Acinetobacter sp. strain NCIB 9871 (33) (see Figs. 6 and 7, which are published as supplemental material on the PNAS web site, www.pnas.org). Two other monooxygenase/regulator pairs with similar gene organization are known that show high homology to both the regulator and monooxygenase components to the MTb locus, one from Dienococcus radiodurans (34) and the other from Streptomyces coelicolor (35). One of the isolated library plasmids containing only the etaR gene was electroporated into MTb and MSm, and the resulting MTb transformants grew as a lawn at 2.5 and 5 μg/ml ETA, indicating that EtaR was solely responsible for ETA resistance. The MSm transformants were able to grow at greater than 200 μg/ml ETA, compared with growth of vector control containing MSm at 50 μg/ml.
To see whether EtaR-mediated repression of EtaA was the cause of ETA resistance we transformed MTb and MSm with pMH29 plasmid constructs containing etaR and etaA separately under the control of a strong constituitive promoter (30). Although we could observe resistance with EtaR constructs in MTb we were not successful in overexpressing EtaA in MTb, suggesting expression of this enzyme is tightly controlled in this organism. MSm overexpressing the putative repressor was found to be ETA-resistant with a measured MIC greater than 62.5 μg/ml on solid media (Fig. 2, lanes c). Although the recombinant MSm were equally susceptible to killing with INH, the bacteria overexpressing EtaA were found to be hypersensitive to ETA with noticeable growth inhibition at 2.5 μg/ml, a level comparable to the normal MIC for MTb (Fig. 2, lanes a). Qualitatively comparable results were obtained when these organisms were treated with ETA S-oxide (although the absolute MIC for the sulfoxide is lower, EtaR conferred resistance and EtaA conferred hypersensitivity). These results suggest that EtaA is directly responsible for thioamide S-oxide oxidative activation and that EtaR modulates expression of this enzyme.
Figure 2.
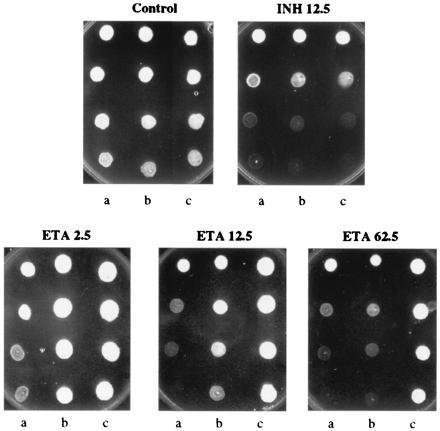
EtaA and EtaR control ETA susceptibility. MSm pMH29 clones expressing EtaA (lanes a), vector control (lanes b), and EtaR (lanes c) are shown spotted in 10-fold decreasing dilutions (from top to bottom) onto 7H11 plates containing the indicated concentration of drugs (in μg/ml). In a microbroth dilution assay the MICs (in μg/ml) for ETA were: lane a, 0.78–1.56; lane b, 50; and lane c, 200 +; for ETA S-oxide: lane a, 0.10–0.20; lane b, 0.78–1.56; and lane c, 6.25–12.5; and for INH: lane a, 1.56–3.13; lane b, 1.56–3.13/6.25, and lane c, 1.56–3.13.
EtaA and EtaR Control ETA Metabolism in Vivo.
To link expression of the EtaA activator more directly with ETA metabolism we examined [14C]ETA conversion by whole cells of the MSm transformants described above over a time-course study as shown in Fig. 3. The EtaA overproducing MSm was found to convert ETA to metabolite 5 much more quickly than vector control (Fig. 3A). Although the EtaR-overproducing strain did appear to affect this conversion less efficiently than the control, the result was not dramatic because MSm normally only weakly activates ETA, consistent with this organism's higher overall MIC for ETA (Fig. 2). These studies directly correlate ETA activation and metabolism with toxicity as measured by MIC. To understand the effect of drug activation we also examined covalent incorporation of [14C]ETA into cellular macromolecules by lysing treated cells and then extensively dialyzing away small molecules. Drug activation was found to correlate directly with incorporation of labeled drug into macromolecules (Fig. 3C).
Figure 3.
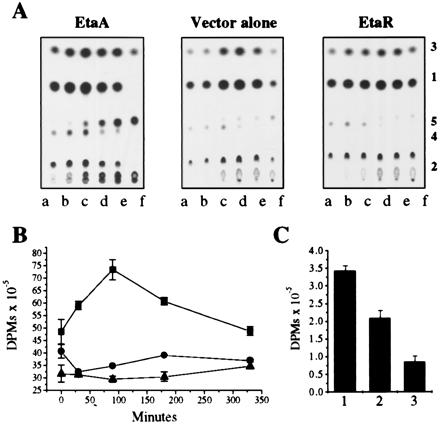
EtaA and EtaR control ETA metabolism. (A) The MSm clones used in Fig. 2 were analyzed for their ability to metabolize [1-14C]ETA. Lanes a–f correspond to sequential filtered culture samples taken at 0, 30, 90, 180, 330, and 900 min, respectively. Metabolites were identified as in Fig. 1 and labeled accordingly. (B) The cell-associated radioactivity for each sequential clone aliquot (1,000 μl) was determined as in Fig. 1B. ■, MSm overexpressing EtaA; ●, wild-type MSm; ▴, MSm overexpressing EtaR. (C) Macromolecule-associated radioactivity was calculated by resuspending microcentrifuged 900-min aliquots (400 μl) in PBS, bead beating (3 × 45 sec), and extensively dialyzing the total lysates to remove molecules smaller than 10 kDa before scintillation analysis. Column 1, MSm overexpressing EtaA; column 2, wild-type MSm; column 3, MSm overexpressing EtaR.
Drug activation, and formation of metabolite 5, also was found to depend on the presence of molecular oxygen, consistent with a role for an oxygen-dependent activation process involving EtaA. When whole cells of MTb were incubated with radioactive drug under an atmosphere of argon, no ETA metabolism was observed (see Figs. 6 and 7).
EtaA Mutation in MDRTB Patient Isolates Resistant to Thioamides.
ETA is only one example of a thiocarbonyl-containing antituberculosis medication approved for clinical use. Among the second-line tuberculosis therapeutics there are two other such molecules, thiacetazone (11) and thiocarlide (isoxyl) (12) (Fig. 4A) that might be similarly activated by EtaA-catalyzed S-oxidation. To elucidate the clinical relevance of EtaA-mediated resistance to thiocarbonyl-containing drugs as a class we characterized a set of 14 multidrug-resistant isolates from patients in Cape Town, South Africa. These isolates were selected on the basis of thiacetazone resistance and then characterized with respect to ETA resistance. Eleven of 14 of these isolates were found to be ETA cross-resistant. Although none of the patients had been treated with thiocarlide 13/14 of the isolates showed thiocarlide cross-resistance.
Figure 4.
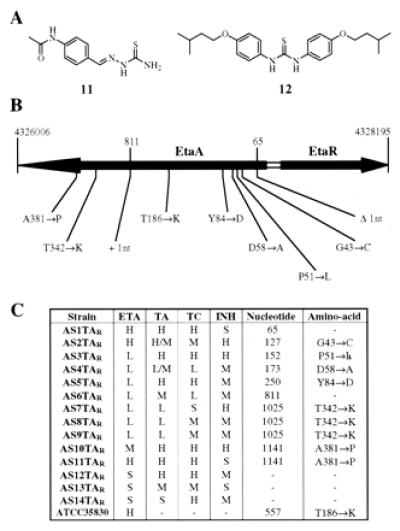
EtaA-associated mutations and cross-resistance in patient isolates from Cape Town, South Africa. (A) Thiacetazone (11) and thiocarlide (12). (B) Map of mutations in EtaA found in patient isolates resistant to ETA and thiacetazone. Chromosome coordinates and gene designations are in reference to the sequenced genome of MTb strain H37Rv. (C) Cross-resistance determination of patient isolates and the associated nucleotide and amino acid alterations observed. Drug susceptibility for ETA is reported as: susceptible (S) if the culture failed to grow at 2.5 μg/ml, low-level resistant (L) if weak growth was observed at 2.5 μg/ml, moderately resistant (M) if strong growth was observed 2.5 μg/ml, and high-level resistant (H) if growth was observed at 10 μg/ml. Drug susceptibility for thiacetazone (TA)/thiocaslide (TC)/INH is reported as susceptible (S) if the culture failed to grow at 0.5 μg/ml, low-level resistant (L) if weak growth was observed at 0.5 μg/ml, moderately resistant (M) if weak growth was observed at 2.0 μg/ml, and high-level resistant (H) if strong growth was observed at 2.0 μg/ml.
To examine at the molecular level the relevance of EtaA-mediated thiocarbonyl activation for this class of compounds we PCR-amplified and sequenced the etaA gene from all 14 multidrug-resistant patient isolates. In addition, we examined an in vitro-generated ETA mono-resistant strain (ATCC 35830). Eleven of 14 clinical isolates had amino acid-altering mutations in EtaA as indicated in Fig. 4B. The nucleotide change at 1025 was found in three isolates, that at 1141 in two isolates. The resulting amino acid alterations tended to occur at positions that were absolutely conserved among other flavin-containing monooxygenases (see Figs. 6 and 7). Along with the single nucleotide changes a 1-nt deletion (65) and addition (811) were found. In the ATCC 35830 ETA mono-resistant strain a nucleotide change at position 557 of EtaA was found. To confirm that these mutations had functional consequences we expressed four of the mutant EtaA proteins in MSm (T186K, T342K, A381P, and Δ1nt65) and demonstrated that the resulting strains were neither ETA-hypersensitive nor converted radio-ETA to the predicted products (data not shown). The three patient isolates where EtaA mutations could not be found corresponded precisely with those that remained fully sensitive to ETA. Thus there is a 100% correspondence between mutation in EtaA and ETA cross-resistance among these thiacetazone-resistant strains.
Discussion
INH (6) has been shown to be activated by KatG in vitro to a variety of products including isonicotinic acid, isonicotinamide, and isonicotinaldehyde (9) [which in vivo is rapidly reduced to 4-pyridylmethanol (10)] (8, 14). The results support the notion that in vivo INH is metabolized by oxidation to an acyl diimide (7), then to a diazonium ion (8) or an isonicotinyl radical that may abstract a hydrogen atom from a suitable donor to form isonicotinaldehyde. Similarly, we postulate that ETA is activated via the corresponding S-oxide (2) to a sulfinate that can form an analogous aldehyde equivalent (an imine) through a radical intermediate (Fig. 5). Hydrolysis of this imine could be followed by reduction of the resulting aldehyde to the observed metabolite (5).
Figure 5.
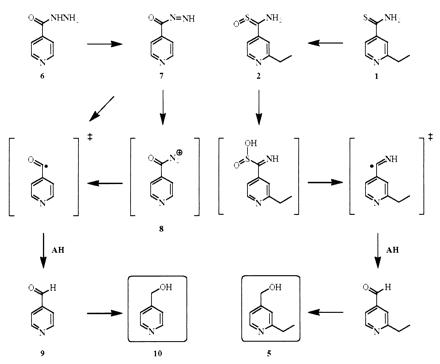
The proposed activation process for thioamide oxidation of ETA.
The mechanistic linkage of the activated form of ETA and INH may explain, in part, the observation that they share a final common target. The striking observation that both drugs give rise to essentially the same final metabolite upon productive activation of the drug further substantiates this common mechanism. Despite this commonality an acyl hydrazide and a thioamide must undergo very different activation processes by discrete enzymes before they converge on an analogous reactive intermediate. The association of KatG with INH activation has been firmly established by a combination of loss of activity studies, laboratory-selected drug-resistant mutants, overexpression, and clinically relevant mutations (7, 36). In this study we establish that EtaA is the analogous enzyme for the activation of ETA and provide similar evidence based on genetic manipulation of the enzyme levels and mutations observed in patient isolates.
EtaA has two closely related homologs (Rv3083 and Rv0565c) encoded within the MTb genome that share almost 50% identity to this monooxygenase (32). It is also a member of a family of 14 more loosely related proteins, the majority of which are probable monoxygenases. In addition, MTb has 20 homologs of cytochrome P450 containing oxygenases, the largest number ever identified within a single bacterial genome (37). The reason for this amazing radiation of oxidative enzymes is not clear but they may have evolved to improve bacterial survival in the face of various xenobiotic substances. In this vein, the ETA susceptibility of this organism may arise from accidental activation by an enzyme intended to aid detoxification.
Thiacetazone (11) has been widely used as a front-line therapeutic in Africa and throughout the developing world because it is extremely inexpensive (38). Although thiocarlide (12) has not been widely used there is renewed interest in this drug and new analogs (39). There is an impressive clinical history of cross-resistance among this set of three second-line therapies (40–46). This cross-resistance suggested a common mechanism of activation of thiocarbonyl containing molecules that might allow the simultaneous acquisition of drug resistance to this class of therapeutic. When we examined the patient isolates from Cape Town for cross-resistance to other thioamides or thioureas we noted that the vast majority of ETA/thiacetazone-resistant isolates were already resistant to thiocarlide, even though these patients were never treated with this drug.
The extensive cross-resistance among these compounds predicts multiple overlapping mechanisms of resistance among clinically used antituberculars: target-associated between INH and ETA and activation-associated between ETA, thiacetazone, and thiocarlide. Such considerations complicate appropriate drug therapy for the treatment of MDRTB, and these results provide an important tool to help understand and quickly characterize the resistance mechanisms operating in a single patient, which may prove vital to a positive outcome.
Supplementary Material
Abbreviations
- ETA
ethionamide
- INH
isoniazid
- MDRTB
multidrug-resistant tuberculosis
- MTb
Mycobacterium tuberculosis
- MIC
minimal inhibitory concentration
- MSm
Mycobacterium smegmatis
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Farmer P, Bayona J, Becerra M, Furin J, Henry C, Hiatt H, Kim J Y, Mitnick C, Nardell E, Shin S. Int J Tuberc Lung Dis. 1998;2:869–876. [PubMed] [Google Scholar]
- 2.Viskum K, Kok-Jensen A. Int J Tuberc Lung Dis. 1997;1:299–301. [PubMed] [Google Scholar]
- 3.Bass J B, Jr, Farer L S, Hopewell P C, O'Brien R, Jacobs R F, Ruben F, Snider D E, Jr, Thornton G. Am J Respir Crit Care Med. 1994;149:1359–1374. doi: 10.1164/ajrccm.149.5.8173779. [DOI] [PubMed] [Google Scholar]
- 4.W. H. O. TB: Groups at Risk, WHO Report on the Tuberculosis Epidemic. Geneva: World Health Organization; 1996. [Google Scholar]
- 5.Crofton J, Chaulet P, Maher D, Grosset J, Harris W, Norman H, Iseman M, Watt B. Guidelines for the Management of Multidrug-Resistant Tuberculosis. Geneva: World Health Organization; 1997. [Google Scholar]
- 6.Barry C E, 3rd, Slayden R A, Sampson A E, Lee R E. Biochem Pharmacol. 2000;59:221–231. doi: 10.1016/s0006-2952(99)00253-1. [DOI] [PubMed] [Google Scholar]
- 7.Slayden R A, Barry C E., 3rd Microbes Infect. 2000;6:1–11. doi: 10.1016/s1286-4579(00)00359-2. [DOI] [PubMed] [Google Scholar]
- 8.Johnsson K, Schultz P G. J Am Chem Soc. 1994;116:7425–7426. [Google Scholar]
- 9.Rozwarski D A, Grant G A, Barton D H R, Jacobs W R, Jr, Sacchettini J C. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um K S, Wilson T, Collins D, Lisle G D, Jacobs W R., Jr Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 11.Wilming M, Johnsson K. Angew Chem Int Ed. 1999;38:2588–2590. doi: 10.1002/(sici)1521-3773(19990903)38:17<2588::aid-anie2588>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 12.Youatt J. Aust J Exp Biol Med Sci. 1960;38:245–250. doi: 10.1038/icb.1960.25. [DOI] [PubMed] [Google Scholar]
- 13.Youatt J. Aust J Chem. 1961;14:308–311. [Google Scholar]
- 14.Youatt J. Am Rev Respir Dis. 1969;99:729–749. doi: 10.1164/arrd.1969.99.5.729. [DOI] [PubMed] [Google Scholar]
- 15.Musser J M. Clin Microbiol Rev. 1995;8:496–514. doi: 10.1128/cmr.8.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rist N. Adv Tuberc Res. 1960;10:69–126. [PubMed] [Google Scholar]
- 17.Johnsson K, King D S, Schultz P G. J Am Chem Soc. 1995;117:5009–5010. [Google Scholar]
- 18.Bieder A, Brunel P, Roquet-Ghaye J, Kries B. Rev Fra Etud Clin Biol. 1966;11:419–423. [PubMed] [Google Scholar]
- 19.Johnston J P, Kane P O, Kibby M R. J Pharm Pharmacol. 1967;19:1–9. doi: 10.1111/j.2042-7158.1967.tb07986.x. [DOI] [PubMed] [Google Scholar]
- 20.Grunert M, Iwainsky H. Arzneim Forsch. 1967;17:411–415. [PubMed] [Google Scholar]
- 21.Prema K, Gopinthan K P. J Indian Inst Sci. 1976;58:16–27. [Google Scholar]
- 22.Bonicke R. Beitr Klin Erforsch Tububerk Lungenkrankh. 1965;132:311–314. [PubMed] [Google Scholar]
- 23.Stevens G J, Hitchcock K, Wang Y K, Coppola G M, Versace R W, Chin J A, Shapiro M, Suwanrumpha S, Mangold B L K. Chem Res Toxicol. 1997;10:733–741. doi: 10.1021/tx9700230. [DOI] [PubMed] [Google Scholar]
- 24.Kucherova N F, Khomutov R M, Budovskii E I, Evadakov V P, Kochetkov N K. Zhurnal Obshchei Khimii. 1959;29:915–919. [Google Scholar]
- 25.Gutekunst G O, Gray H L. J Am Chem Soc. 1922;44:1741–1746. [Google Scholar]
- 26.Lawrie K W M, Novellie C E A, Saunders D, Coates W J. J Labeled Compounds Radiopharm. 1995;36:891–898. [Google Scholar]
- 27.Sunay U B, Talbot K, Prasad K, Lee G, Jones L. J Labeled Compound Radiopharm. 1995;36:529–536. [Google Scholar]
- 28.Meinert M C, Nunez H A, Byerrum R U. J Labeled Compounds Radiopharm. 1978;14:893–896. [Google Scholar]
- 29.Walter W, Curts J. Chem Ber. 1960;93:1511–1515. [Google Scholar]
- 30.Mdluli K, Sherman D R, Hickey M J, Kreiswirth B N, Morris S, Stover C K, Barry C E., 3rd J Infect Dis. 1996;174:1085–1090. doi: 10.1093/infdis/174.5.1085. [DOI] [PubMed] [Google Scholar]
- 31.George K M, Yuan Y, Sherman D R, Barry C E., 3rd J Biol Chem. 1995;270:27292–27298. doi: 10.1074/jbc.270.45.27292. [DOI] [PubMed] [Google Scholar]
- 32.Cole S T, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon S V, Eiglmeier K, Gas S, Barry C E, 3rd, et al. Nature (London) 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y C, Peoples O P, Walsh C T. J Bacteriol. 1988;170:781–789. doi: 10.1128/jb.170.2.781-789.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White O, Eisen J A, Heidelberg J F, Hickey E K, Peterson J D, Dodson R J, Haft D H, Gwinn M L, Nelson W C, Richardson D L, et al. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redenbach M, Kieser H M, Denapaite D, Eichner A, Cullum J, Kinashi H, Hopwood D A. Mol Microbiol. 1996;21:77–96. doi: 10.1046/j.1365-2958.1996.6191336.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Heym B, Allen B, Young D, Cole S. Nature (London) 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 37.Nelson D R. Arch Biochem Biophys. 1999;369:1–10. doi: 10.1006/abbi.1999.1352. [DOI] [PubMed] [Google Scholar]
- 38.Nunn P, Porter J, Winstanley P. Trans R Soc Trop Med Hyg. 1993;87:578–582. doi: 10.1016/0035-9203(93)90096-9. [DOI] [PubMed] [Google Scholar]
- 39.Phetsuksiri B, Baulard A R, Cooper A M, Minnikin D E, Douglas J D, Besra G S, Brennan P J. Antimicrob Agents Chemother. 1999;43:1042–1051. doi: 10.1128/aac.43.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muzikravic T. Antibiot Chemother. 1970;16:177–181. [PubMed] [Google Scholar]
- 41.Sojkova M, Tousek J, Trnka L. Praxis Pneumol Vereinigt Tuberk. 1965;19:522–527. [PubMed] [Google Scholar]
- 42.Murohashi, Yanagisawa Acta Tuberc Pneumol Belg. 1963;54:35–40. [Google Scholar]
- 43.Tskamura M. Kekkaku. 1962;37:103–113. [PubMed] [Google Scholar]
- 44.Afanasieva Y P, Mkrtchyan S V. Probl Tuberk. 1969;47:66–99. [Google Scholar]
- 45.Verbist L. Antimicrob Agents Chemother. 1965;5:298–305. [PubMed] [Google Scholar]
- 46.Konopka E, Gisi T, Eisman P. Proc Soc Exp Biol. 1955;89:388–398. doi: 10.3181/00379727-89-21819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.