

Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative condition caused by expansions of more than 35 uninterrupted CAG repeats in exon 1 of the huntingtin gene. The CAG repeats in HD and the other seven known diseases caused by CAG codon expansions are translated into long polyglutamine tracts that confer a deleterious gain of function on the mutant proteins. Intraneuronal inclusions comprising aggregates of the relevant mutant proteins are found in the brains of patients with HD and related diseases. It is crucial to determine whether the formation of inclusions is directly pathogenic, because a number of studies have suggested that aggregates may be epiphenomena or even protective. Here, we show that fragments of the bacterial chaperone GroEL and the full-length yeast heat shock protein Hsp104 reduce both aggregate formation and cell death in mammalian cell models of HD, consistent with a causal link between aggregation and pathology.
Huntington's disease (HD) is an autosomal dominant neurodegenerative condition associated with abnormal movements, cognitive deterioration, and psychiatric symptoms. The causative mutation is a (CAG)n trinucleotide-repeat expansion of more than 35 repeats, which is translated into an abnormally long polyglutamine tract in the huntingtin protein (reviewed in refs. 1 and 2).
HD is a member of a family of neurodegenerative diseases caused by CAG/polyglutamine expansions, which include spinobulbar muscular atrophy (SBMA), spinocerebellar ataxias (SCA) types 1, 2, 3, 6, and 7, and dentatorubral–pallidoluysian atrophy (DRPLA). All diseases are dominantly inherited (except for SBMA, which is X-linked). In all cases, age at onset correlates inversely with repeat number (reviewed in ref. 2). The polyglutamine expansion mutation causes disease by conferring a novel deleterious function on the mutant protein, and the severity correlates with increasing CAG repeat number and expression levels in transgenic mice (3) and in cell culture models (4).
Although each of these diseases is associated with specific regions of neurodegeneration (which, in some cases, overlap), they probably are caused by similar pathological processes. A hallmark of many of these diseases, including HD (5), SBMA (6), DRPLA (7), and SCA types 1 (8), 2 (9), 3 (10), 6 (11), and 7 (12), is the development of intracellular protein aggregates (inclusions) in the vulnerable neurons. A pathological role for inclusions is suggested by the correlation of the number of inclusions in the cortex of HD patients with CAG repeat number, which reflects disease severity (13). Inclusion formation precedes neurological dysfunction in some HD transgenic mice (14) and is associated with predisposition to cell death in cell culture models of HD (15–17), DRPLA (18), SBMA (19), SCA3 (10), and SCA6 (11).
The hypothesis that inclusions have a direct pathogenic role in these diseases has been challenged by experiments reporting a dissociation between cell death and inclusion formation in primary cell cultures; inhibition of ubiquitination was associated with decreased aggregate formation but more cell death (20). These findings were not straightforward, because inhibition of ubiquitination also increased apoptosis in cells expressing wild-type (wt) huntingtin constructs and others have suggested that these data still may be compatible with a pathogenic role for huntingtin polymerization (21). Klement and colleagues (22, 23) suggested that inclusions may not be pathogenic, because deletion of the self-association domain from a SCA1 transgene with expanded repeats prevented the inclusion formation seen in mice expressing full-length mutant SCA1 transgenes, but both mouse models developed a SCA-like phenotype. Perutz (21) argued that this conclusion was unwarranted, because deletion of these 122 residues would turn the protein into a random coil. Thus, the experiment shows that Purkinje cell expression of denatured, truncated ataxin-1 gives rise to ataxia. One cannot argue that this effect was related to the polyglutamine expansion, because no data were presented for mice expressing wt polyglutamine lengths in ataxin-1 with deletion of the self-association domain. Recently, Cummings et al. (24) showed that loss of function of the E6-AP ubiquitin ligase reduced the formation of nuclear inclusions but accelerated polyglutamine-induced pathology in SCA1 mice. Although these data suggest that large, visible inclusions may not be required for cell death, the authors considered other possibilities that are compatible with a pathological role for inclusions. The loss of E6-AP activity may not have had a direct effect on the ubiquitination and clearance of ataxin-1 (24) but may have increased the half-lives of many other cellular proteins, which, at abnormally high steady-state levels, may have enhanced the cellular sensitivity to the SCA1 mutation (or aggregates).
Polyglutamine (polyQ) diseases have been studied extensively by using exon 1 fragments of huntingtin, because large fragments of the HD, SCA3, and DRPLA gene products do not induce inclusion formation or cell death in cell culture models (10, 15, 16, 19). A small N-terminal polyQ-containing part(s) of huntingtin is found in inclusions in vivo (5), but the exact nature and role of this fragment(s) is unclear. Although exon 1 models may not be specific for HD, they are powerful tools for studying inclusion formation in relation to cell death/dysfunction in cultured cells and transgenic mice. We have used cell models of HD comprising an exon 1 fragment with varying CAG repeat lengths tagged at the N terminus with enhanced green fluorescent protein (EGFP) (4, 17). This model, like other published cell models (15, 16, 19), shows many of the features observed in vivo: constructs with repeat lengths in the normal size range (23 glutamines) do not aggregate, but constructs with 43 or more glutamines do aggregate and enhance cell death, and these phenotypes correlate with the duration and levels of transgene expression (4, 17). The deleterious effects of polyglutamine mutations have been observed in both neuronal (e.g., PC12) and nonneuronal (e.g., COS-7) cell lines, consistent with the notion that although neuronal cell death and neurological symptoms are the cardinal features of these diseases, the mutations also can affect nonneuronal cells in humans and in mouse models (25–28).
In this study we show that fragments of the bacterial chaperone GroEL and the full-length yeast heat shock protein (Hsp) Hsp104 reduce both aggregate formation and cell death in mammalian cell models of HD, consistent with a direct, causal link between aggregation and cell dysfunction/death. In addition, we show that a monomer comprising GroEL residues 191–345 (29) and an artificial heptamer formed by seven copies of a GroEL minichaperone (residues 191–376) have chaperone activity in mammalian cells. Thus, we resolve another important debate (30, 31) by providing direct evidence that the large, central cavity of GroEL is not essential for its aggregate-reducing activity in vivo. Aggregate formation and cell death also were reduced by the K620T mutant of Hsp104, which impairs its oligomerization in vitro (32). Thus, efficient oligomerization may not be an essential requirement for all Hsp104 functions, in a fashion analogous to the GroEL 191–345 monomer.
Materials and Methods
Plasmid Construction.
MC7 was constructed by inserting a minichaperone GroEL into GroES in the pRSETA vector (33). The DNA sequence encoding a part of the mobile loop of GroES (residues 16–33) was removed by PCR, as described (34), using the oligonucleotides 5′-TCC GGC TCT GCA GCG G-3′ and 5′-TCC AGA GCC AGT TTC AAC TTC TTT ACG C-3′, creating a unique BamHI site (bold characters) and the vector pRSETA-GroESΔloop. The groEL minichaperone gene [corresponding to the apical domain of GroEL, residues 191–376 (29)] was amplified by PCR by using primers containing a BamHI site (underlined) 5′-TTC GGA TCC GAA GGT ATG CAG TTC GAC C-3′ and 5′-GTT GGA TCC AAC GCC GCC TGC CAG TTT C-3′ and cloned into the unique BamHI site of pRSETA-GroESΔloop vector, thus inserting the minichaperone GroEL(191–376) in-frame into the GroESΔloop sequence.
We PCR-amplified the genes encoding MC7, GroEL wt (full-length groEL), Hsp104 wt, and Hsp104 K620T from bacterial expression vectors (pRSETA for the MC7 and GroEL constructs and pGal for the Hsp104 constructs) and cloned them into the mammalian expression vector pCDNA 3.1/His B (Invitrogen). To allow cloning into the corresponding sites in the pCDNA vector, the primers for the GroEL constructs had Bsu36I and EcoRI restriction sites (for GroEL wt and MC7) incorporated in them and the primers for the Hsp104 constructs had BamHI and NotI restriction sites incorporated in them. The primers were as follows: GroEL wt Bsu36I, 5′-ACGTCCTAAGGATATGGCAGCTAAAGACGTAAAATTC-3′; GroEL wt EcoRI, 5′-ACGTGGATTCTTACATCATGCCGCCCATGCCACC-3′; MC7 Bsu36I, 5′-ACGTCCTAAGGATATGAATATTCGTCCATTGCATGAT-3′; MC7 EcoRI, 5′-ACGTGAATTCTTACGCTTCAACAATTGCCAGAAT-3′; Hsp104 BamHI, 5′-ACGTGGATCCAATGAACGACCAAACGCAATTTAC-3′; Hsp104 NotI, 5′-ACGTGCGGCCGCTTAATCTAGGTCATCATCAATTTCC-3′.
ApGroEL (corresponding to the apical domain of GroEL residues 191–345) was released from pRSETA (29) with EcoRI and BamHI (New England Biolabs) and subcloned into the respective restriction sites in the pCDNA 3.1/His B. All constructs were validated by sequencing.
The HD exon 1 constructs in pEGFP-C1 constructs have been described (4, 17).
Cell Culture and Transfection Experiments.
COS-7 and PC12 cells were cultured, transfected, and fixed as described (4, 17). We used a 3:1 or 5:1 molar ratio of pCDNA 3.1/His B constructs to HD exon 1 construct DNA to ensure that all cells expressing HD exon 1 constructs also expressed the appropriate pCDNA 3.1/His B construct. In all such experiments, we used a total of 2 μg of DNA per 3.5-cm dish and kept the amount of the HD exon 1 construct constant in test and control conditions. We analyzed between 300 and 600 EGFP-expressing cells per slide (blinded) in multiple, randomly chosen visual fields. The proportion of HD exon 1-expressing cells with one or more intracellular inclusions was used as a measure of inclusion formation, following Cummings et al. (35). Odds ratios and P values were determined by unconditional logistical regression analysis, using the general log-linear analysis option of spss 9 software (SPSS, Chicago).
For Western blotting, cell lysates were electrophoresed on 15% denaturing gels, and an anti-His6 mouse mAb (CLONTECH) diluted 1:2,500 was used as the primary antibody. Blots were probed with horseradish peroxidase-labeled anti-mouse antibody (Amersham Pharmacia) and bands were detected with the ECL (enhanced chemiluminescent) detection reagent (Amersham Pharmacia).
Results
Bacterial and Yeast Chaperones Reduce Polyglutamine Aggregation.
In this study we have tested the effects of five molecular chaperone constructs on inclusion formation and cell death in cell models of HD. These included wt yeast Hsp104 (Hsp104 wt), which reduces aggregation when overexpressed in yeast models of HD (36). No close mammalian homologues of this protein are known. Hsp104 wt was compared with the K620T mutation (Hsp104 K620T) in the second nucleotide-binding domain (32). This mutation impairs oligomerization in vitro, with only a partial reduction in ATPase activity (32). We examined the bacterial chaperonin GroEL (GroEL wt), a large complex comprising 14 identical, ≈57.5-kDa subunits organized in a two-ring structure. We tested whether the large, central cavity of GroEL was essential for its chaperone activity by studying a monomeric minichaperone corresponding to part of its apical polypeptide-binding domain (residues 191–345) (apGroEL) (29). We also investigated the effect of placing seven copies of a GroEL minichaperone (residues 191–376) in a ring, which we called MC7. This mimics the organization of one of the rings of wt GroEL. MC7 was made by inserting GroEL residues 191–376 in the place of part of the highly mobile loop (residues 16–33) of the bacterial cochaperonin GroES (J. Chatellier, Fergal Hill, and A.R.F., unpublished results). GroES forms a stable structure of seven ≈10.4-kDa subunits (37). When analyzed by both analytical size-exclusion chromatography and analytical ultracentrifugation, recombinant MC7 formed heptamers comprising seven 30-kDa subunits, and electron microscopic studies of MC7 revealed a diameter similar to that of GroEL wt (O. Llorca, J. Chatellier, J.-L. Carrascosa, and A.R.F., unpublished data). The yeast and bacterial chaperone constructs described above were expressed from the mammalian expression vector pcDNA 3.1/HisB; proteins of appropriate sizes from lysates of transfected cells were detected by Western blotting by using an anti-His6 antibody (data not shown).
The functions of GroEL wt, apGroEL, MC7, Hsp104 wt, and Hsp104 K620T were investigated by cotransfecting each of the test constructs into either COS-7 or PC12 (rat pheochromocytoma) cell lines with constructs expressing EGFP-tagged–exon 1 fragments of huntingtin with 53 or 74 glutamines (EGFP-HDQ53 and EGFP-HDQ74, respectively) (4, 17). The empty pFLAG expression vector and a J domain-deleted form of the human Hsp40 homologue, HDJ2 (called Δ450) (17, 35), were used as controls. A 3:1 or 5:1 molar ratio of chaperone (and control plasmid) to EGFP-HD exon 1 constructs was used to ensure that all EGFP-expressing cells also expressed the chaperone or control plasmid (38). Data from our experiments (each performed in triplicate on two to four occasions) are expressed as odds ratios, which are the proportions of EGFP-expressing cells with inclusions divided by proportions of EGFP-expressing cells without inclusions when cotransfected with the chaperones, divided by the same ratio, when cotransfected with the control plasmids. Odds ratios were considered to be the most appropriate summary statistic, because the percentage of cells with inclusions under specified conditions varied between experiments on different days, whereas the relative change in the proportion of cells with inclusions induced by an experimental perturbation is expected to be more consistent (17, 38).
We tested the effects of these chaperones at various time points (see Fig. 1). In general, we analyzed the EGFP-HDQ74 construct at 48 h, where 40–50% of EGFP-positive cells had inclusions and 40–45% of EGFP-positive cells had fragmented nuclei when cotransfected with pFLAG. EGFP-HDQ53 was analyzed in the greatest detail at 72 h to allow sufficient formation of inclusions (30–40% of EGFP-positive cells had inclusions and 24–33% cells had fragmented nuclei when cotransfected with pFLAG). apGroEL, MC7, Hsp104 wt, and Hsp104 K620T significantly reduced the proportions of EGFP-positive cells with aggregates in both COS-7 and PC12 lines, compared with the control constructs (Fig. 1). The most pronounced effects were observed with MC7, where aggregation was reduced by 50% in PC12 cells and by 30% in COS-7 cells. apGroEL consistently and significantly reduced aggregation caused by EGFP-HDQ53 and EGFP-HDQ74, and effects were seen in both cell lines. GroEL wt significantly reduced inclusions in PC12 cells, but the reduction was not significant in COS-7 cells. Δ450 showed no difference in inclusion formation compared with pFLAG (Fig. 1). We previously have tested a large number of different proteins for their ability to modify aggregation in these cell lines, and virtually all of these have no effect or they enhance aggregation (17, 38). This confirms that the effects we have reported here are specific.
Figure 1.
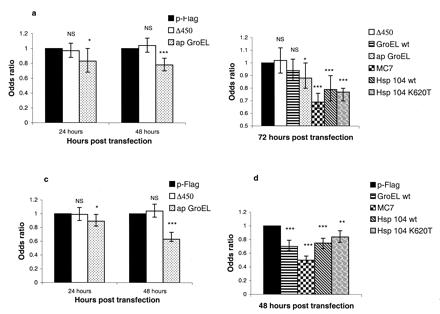
Suppression of inclusion formation by apGroEL, MC7, Hsp104 wt, and Hsp104 K620T in COS-7 (a and b) and PC12 (c and d) cells. In all experiments, the odds ratios are derived from 2–4 independent experiments, each done in triplicate, where we have compared test constructs with pFLAG. (Error bars = 95% confidence intervals for the odds ratios.) (a) COS-7 cells cotransfected with EGFP-HDQ74 and apGroEL and control plasmids. (b) COS-7 cells cotransfected with EGFP-HDQ53 and all test and control plasmids. (c) PC12 cells cotransfected with EGFP-HD74 and apGroEL and control plasmids. (d) PC12 cells cotransfected with EGFP-HD74 and all test and control plasmids (except for apGroEL, which is shown in c). *, P < 0.05; **, P < 0.001; ***, P < 0.0001; NS = P > 0.05.
Bacterial and Yeast Chaperones Reduce Cell Death.
Nuclear fragmentation can be assessed accurately in polyglutamine disease models in COS-7 cells (17, 39, 40). Because PC12 cells transiently expressing EGFP-HDQ74 appear to have less obvious nuclear fragmentation, it is difficult to quantify this phenotype objectively in this cell line. We scored the proportion of EGFP-positive cells with fragmented nuclei in COS-7 cells to determine whether a reduction in inclusion formation was associated with a reduction in cell death (Fig. 2). GroEL wt, apGroEL, MC7, and both Hsp104 wt and Hsp104 K620T reduced the proportion of EGFP-positive cells with fragmented vs. normal nuclei compared with the control plasmids. Δ450 did not modify cell death when compared with pFLAG (Fig. 2).
Figure 2.
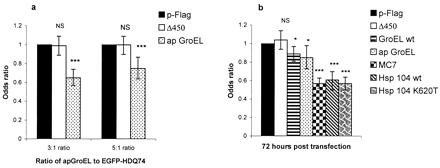
Reduction of nuclear fragmentation in COS-7 cells after coexpression of EGFP-HDQ74 (a) and EGFP-HDQ53 (b) with apGroEL, MC7, Hsp104 wt, and Hsp104 K620T. Cells were analyzed after 48 h (a) and 72 h (b). In all experiments, the odds ratios are derived from 2–4 independent experiments, each done in triplicate, where we have compared test constructs with pFLAG. (Error bars = 95% confidence intervals for the odds ratios.) *, P < 0.05; *, P < 0.001; *, P < 0.0001; NS = P > 0.05.
We performed immunocytochemical analyses in COS-7 cells coexpressing GroEL wt, apGroEL, MC7, or Hsp104 and EGFP-huntingtin mutant exon 1 fragments, because previous studies have shown that some heat shock proteins colocalize with polyglutamine inclusions (17, 35, 41). The chaperones used here did not colocalize with these polyglutamine inclusions (data not shown).
Discussion
Our data strengthen the case for a pathological role for aggregates in polyglutamine diseases, because our panel of bacterial and yeast chaperones reduced both aggregate formation and cell death in cell culture models of HD. We previously have reported that EGFP-HDQ74-expressing COS-7 cells with inclusions showed significantly more nuclear fragmentation at 24, 48, and 72 h posttransfection compared with either EGFP-HDQ23-expressing cells or EGFP-HDQ74-expressing cells without inclusions, which had similar death rates (17). The same association between inclusion formation and cell death was observed when we generated intracellular aggregates with EGFP fused to 19–35 alanines and compared these cells with those expressing native EGFP or EGFP fused to 7 alanines, which did not aggregate (39). Previous studies have shown that the Hsp40 homologue HDJ-1 (41) and the polyglutamine-binding peptide QBP1 (40) reduce both inclusion formation and cell death in tissue culture models. It is possible that HDJ-1 modifies cell death processes via pathways independent of its effects on aggregation formation, because it is a cochaperone of Hsp70, which may be able to directly modulate cell death pathways (42). Nagai et al. (40) suggested that QBP1 may exert its effect on cell death by inhibiting interactions of polyglutamine proteins with other molecules and that its aggregate-reducing effect consequently may be an epiphenomenon. These caveats to the link between aggregation and death are extremely unlikely with the panel of chaperones that we tested, because these bacterial and yeast proteins are unlikely to impact directly on mammalian death pathways (independent of aggregation) and apGroEL, MC7 (J. Chatellier, Fergal Hill, and A.R.F., unpublished results), and Hsp104 all have chaperone activity in vitro (29, 32).
It is notable that bacterial and yeast chaperones can function in mammalian cells, and the data from apGroEL and MC7 inform the important debate relating to whether the large, central cavity of GroEL is essential for all aspects of its activity (30, 31). GroEL wt did not appear to be as efficient as the apGroEL and MC7 minichaperones, presumably because its activity in vitro and in vivo is GroES-dependent (29), in contrast to the minichaperones. This report of minichaperones directly reducing aggregation in mammalian cells suggests that the large, central cavity of GroEL is not essential for all of its activities and that the apical fragment can act independently of the microenvironment fostered by the complex structure of native multimeric GroEL, as demonstrated previously in Escherichia coli and bacteriophage λ (33). Our findings that Hsp104 can act in mammalian cells complement the observations that this protein reduces polyglutamine aggregation and cell death in a Caenorhabditis elegans model that were published while we were writing this paper (43). Aggregate formation and cell death also were reduced by the K620T mutant of Hsp104, which impairs its oligomerization in vitro (32). Although transient oligomerization may occur in vivo, Hsp104 may not require efficient oligomerization for all of its functions, in a manner analogous to the GroEL(191–345) monomer.
Although our results support a pathological role for inclusions, they are not necessarily incompatible with previous data (20, 22–24), because it is possible that mutant forms of these proteins also may be toxic in the absence of visible aggregation. Because these chaperones reduce cell death, they may provide the basis for therapeutic strategies not only for polyglutamine diseases, but also for more common neurodegenerative conditions associated with protein aggregation, including forms of motor neuron disease, Parkinson's disease, and Alzheimer's disease.
Acknowledgments
We thank Drs. Chris Cummings, Huda Zoghbi, and Susan Lindquist for the HDJ-2/HSDJ and Hsp104 constructs. We are grateful to Evan Reid, Rainer Duden, Andreas Wyttenbach, and Jina Swartz for helpful discussion. J. Carmichael is an Action Research Training Fellow and is grateful for a Sackler Studentship, and D.C.R. is a Glaxo Wellcome Research Fellow. J. Chatellier was a Marie Curie EU fellow. C.M. and A.W. are grateful for a grant from the National Foundation for Cancer Research (USA), and A.R.F. is grateful for a grant from the Wellcome Trust.
Abbreviations
- HD
Huntington's disease
- SBMA
spinobulbar muscular atrophy
- SCA
spinocerebellar ataxias
- DRPLA
dentatorubral–pallidoluysian atrophy
- EGFP
enhanced green fluorescent protein
- Hsp
heat shock protein
- wt
wild type
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170280697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170280697
References
- 1.The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Rubinsztein D C, Wyttenbach A, Rankin J. J Med Genet. 1999;36:265–270. [PMC free article] [PubMed] [Google Scholar]
- 3.Davies S W, Turmaine M, Cozens B A, DiFiglia M, Sharp A H, Ross C A, Scherzinger E, Wanker E E, Mangiarini L, Bates G P. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 4.Narain Y, Wyttenbach A, Rankin J, Furlong R A, Rubinsztein D C. J Med Genet. 1999;36:739–746. doi: 10.1136/jmg.36.10.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiFiglia M, Sapp E, Chase K O, Davies S W, Bates G P, Vonsattel J P, Aronin N. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Miwa S, Kobayashi Y, Merry D E, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck K H, Sobue G. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- 7.Igarashi S, Koide R, Shimohata T, Yamada M, Hayashi Y, Takano H, Date H, Oyake M, Sato T, Sato A, et al. Nat Genet. 1998;18:111–117. doi: 10.1038/ng0298-111. [DOI] [PubMed] [Google Scholar]
- 8.Skinner P J, Koshy B T, Cummings C J, Klement I A, Helin K, Servadio A, Zoghbi H Y, Orr H T. Nature (London) 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 9.Koyano S, Uchihara T, Fujigasaki H, Yagishata S, Iwabuchi K. Neurosci Lett. 1999;273:117–120. doi: 10.1016/s0304-3940(99)00656-4. [DOI] [PubMed] [Google Scholar]
- 10.Paulson H L, Perez M K, Trottier Y, Trojanowski J Q, Subramony S H, Das S S, Vig P, Mandel J L, Fischbeck K H, Pittman R N. Neuron. 1997;19:333–334. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 11.Ishikawa K, Fujigasaki H, Saegusa H, Ohwada K, Fujita T, Iwamoto H, Komatsuzaki Y, Toru S, Toriyama H, Watanabe M, et al. Hum Mol Genet. 1999;8:1185–1193. doi: 10.1093/hmg/8.7.1185. [DOI] [PubMed] [Google Scholar]
- 12.Holmberg M, Duyckaerts C, Durr A, Cancel G, Gourfinkel-An I, Damier P, Faucheux B, Trottier Y, Hirsch E C, Agid Y, et al. Hum Mol Genet. 1998;7:913–918. doi: 10.1093/hmg/7.5.913. [DOI] [PubMed] [Google Scholar]
- 13.Becher M W, Kotzuk J A, Sharp A H, Davies S W, Bates G P, Price D L, Ross C A. Neurobiol Dis. 1997;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 14.Davies S W, Turmaine M, Cozens B A, DiFiglia M, Sharp A H, Ross C A, Scherzinger E, Wanker E E, Mangiarini L, Bates G P. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 15.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim S U, et al. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 16.Cooper J K, Schilling G, Peters M F, Herring W J, Sharp A H, Kaminsky Z, Masone J, Khan F A, Delanoy M, Borchelt D R, et al. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 17.Wyttenbach A, Carmichael J, Swartz J, Furlong R A, Narain Y, Rankin J, Rubinsztein D C. Proc Natl Acad Sci USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato A, Shimohata T, Koide R, Takano H, Sato T, Oyake M, Igarashi S, Tanaka K, Inuzuka T, Nawa H, et al. Hum Mol Genet. 1999;8:997–1006. doi: 10.1093/hmg/8.6.997. [DOI] [PubMed] [Google Scholar]
- 19.Ellerby L M, Hackam A S, Propp S S, Ellerby H M, Rabizadeh S, Cashman N R, Trifiro M A, Pinsky L, Wellington C L, Salvesen G S, et al. J Neurochem. 1999;2:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 20.Sadou F, Finkbeiner S, Devys D, Greenberg M E. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 21.Perutz M F. Trends Biochem Sci. 1999;24:58–63. doi: 10.1016/s0968-0004(98)01350-4. [DOI] [PubMed] [Google Scholar]
- 22.Klement I A, Skinner P J, Kaytor M D, Yi H, Hersch S M, Clark H B, Zoghbi H Y, Orr H T. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 23.Orr, H. T., Skinner, P. A., Klement, C. J., Cummings, C. J. & Zoghbi, H. J. (1998) Am. J. Hum. Genet., Suppl., A8.
- 24.Cummings C J, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr H T, Beaudet A L, Zoghbi H Y. Neuron. 1999;24:879–892. doi: 10.1016/s0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- 25.Sawa A, Wiegand G W, Cooper J, Margolis R L, Sharp A H, Lawler J F, Jr, Greenamyre J T, Snyder S H, Ross C A. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 26.Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, Wanker E E, Doherty P, Davies S W, Bates G P. Hum Mol Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- 27.Arenas J, Campos Y, Ribacoba R, Martin M A, Rubio J C, Ablanedo P, Cabello A. Ann Neurol. 1998;43:397–400. doi: 10.1002/ana.410430321. [DOI] [PubMed] [Google Scholar]
- 28.Hurlbert M S, Zhou W, Wasmeier C, Kaddis F G, Hutton J C, Freed C R. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- 29.Zahn R, Buckle A M, Perrett S, Johnson C M, Corrales F J, Golbik R, Fersht A R. Proc Natl Acad Sci USA. 1996;93:15024–15029. doi: 10.1073/pnas.93.26.15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J D, Michelitsch M D, Weissman J S. Proc Natl Acad Sci USA. 1998;95:12163–12168. doi: 10.1073/pnas.95.21.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber F, Keppel F, Georgopoulos C, Hayer-Hartl M K, Hartl F U. Nat Struct Biol. 1998;5:977–985. doi: 10.1038/2952. [DOI] [PubMed] [Google Scholar]
- 32.Schirmer E C, Queitsch C, Kowal A S, Parsell D A, Lindquist S. J Biol Chem. 1998;273:15546–15552. doi: 10.1074/jbc.273.25.15546. [DOI] [PubMed] [Google Scholar]
- 33.Chatellier J, Hill F, Lund P A, Fersht A R. Proc Natl Acad Sci USA. 1998;95:9861–9866. doi: 10.1073/pnas.95.17.9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemsley A, Arnheim N, Toney M D, Cortopassi G, Galas D J. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cummings C J, Mancini M A, Antalffy B, DeFranco D B, Orr H T, Zoghbi H Y. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 36.Krobitsch S, Lindquist S. Proc Natl Acad Sci USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunt J F, Weaver A J, Landry S J, Gierasch L, Deisenhofer J. Nature (London) 1996;379:37–45. doi: 10.1038/379037a0. [DOI] [PubMed] [Google Scholar]
- 38.Furlong R A, Narain Y, Rankin J, Wyttenbach A, Rubinsztein D C. Biochem J. 2000;346:577–581. [PMC free article] [PubMed] [Google Scholar]
- 39.Rankin J, Wyttenbach A, Rubinsztein D C. Biochem J. 2000;348:15–19. [PMC free article] [PubMed] [Google Scholar]
- 40.Nagai Y, Tucker T, Ren H, Kenan D J, Henderson B S, Keene J D, Strittmatter W J, Burke J R. J Biol Chem. 2000;275:10437–10442. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 41.Chai Y, Koppenhafer S L, Bonini N M, Paulson H L. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warrick J M, Chan H Y, Gray-Board G L, Chai Y, Paulson H L, Bonini N M. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 43.Satyal S H, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer J M, Morimoto R I. Proc Natl Acad Sci USA. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]