

Abstract
Production of amyloid-β protein (Aβ) is initiated by a β-secretase that cleaves the Aβ precursor protein (APP) at the N terminus of Aβ (the β site). A recently identified aspartyl protease, BACE, cleaves the β site and at residue 11 within the Aβ region of APP. Here we show that BACE2, a BACE homolog, cleaves at the β site and more efficiently at a different site within Aβ. The Flemish missense mutation of APP, implicated in a form of familial Alzheimer's disease, is adjacent to this latter site and markedly increases Aβ production by BACE2 but not by BACE. BACE and BACE2 respond identically to conservative β-site mutations, and alteration of a common active site Arg inhibits β-site cleavage but not cleavage within Aβ by both enzymes. These data suggest that BACE2 contributes to Aβ production in individuals bearing the Flemish mutation, and that selective inhibition of these highly similar proteases may be feasible and therapeutically advantageous.
The amyloid-β protein (Aβ) is the principal component of the senile plaques characteristic of Alzheimer's disease (AD), and multiple lines of evidence have implicated cerebral accumulation of Aβ in AD pathogenesis (1, 2). Aβ is produced from the Aβ precursor protein (APP) by two proteolytic events. A β-secretase activity cleaves APP at the N terminus of Aβ (β site) between amino acids Met-671 and Asp-672 (using the numbering of the 770-aa isoform of APP). Cleavage at the β site yields a membrane-associated APP fragment of 99 aa (C99). A second site within the transmembrane domain of C99 (γ site) can then be cleaved by a γ-secretase to release Aβ, a peptide of 39–42 aa. APP can alternatively be cleaved within its Aβ region, predominately at the α-secretase cleavage site of APP, to produce a C-terminal APP fragment of 83 aa (C83), which can also be further cleaved by γ-secretase to produce a small secreted peptide, p3.
A number of missense mutations in APP have been implicated in forms of early-onset familial AD. All of these are at or near one of the canonical cleavage sites of APP. Thus, the Swedish double mutation (K670N/M671L) is immediately adjacent to the β-cleavage site and increases the efficiency of β-secretase activity, resulting in more total Aβ (3). Any of three mutations at APP residue 717, near the γ site, increases the proportion of a more amyloidogenic 42-aa form of Aβ [Aβ(1–42)] relative to the more common 40-residue form [A(β1–40)] (4–7).
Two additional mutations of APP have been described which are close but not adjacent to the α site. A mutation (A692G, Aβ residue 21) in a Flemish family and a mutation (E693Q, Aβ residue 22) in a Dutch family each have been implicated in distinct forms of familial AD (8–10). The Flemish mutation, in particular, presents as a syndrome of repetitive intracerebral hemorrhages or as an AD-type dementia. The neuropathological findings include senile plaques in the cortex and hippocampus, and usually multiple amyloid deposits in the walls of cerebral microvessels (8, 11, 12).
Recently, a membrane-associated aspartyl protease, BACE (also called β-secretase or Asp2) has been shown to exhibit properties expected of a β-secretase (13–16). This enzyme cleaves APP at its β site and between Tyr-10 and Glu-11 of the Aβ region with comparable efficiency (15). Aβ fragments cleaved at this latter site have been observed in amyloid plaques in AD and in media of APP-transfected HEK293 human embryonic kidney cells (17–19). Several groups also observed the presence in the database of an additional aspartyl protease, BACE2 (also called Asp1), a close homolog of BACE (hereafter referred to as BACE1) (16, 20).
We show here that BACE2 cleaves APP at its β site and more efficiently at sites within the Aβ region of APP, after Phe-19 and Phe-20 of Aβ. These internal Aβ sites are adjacent to the Flemish APP mutation at residue 21, and this mutation markedly increases the proportion of β-site cleavage product generated by BACE2. We find that conservative β-site mutations of APP that either increase (the Swedish mutation) or inhibit (M671V) β-secretase activity affect BACE1 and BACE2 activity similarly, and that BACE2, like BACE1, proteolyzes APP maximally at acidic pH. Moreover, alteration of a single Arg common to both enzymes blocks their ability to cleave at the β site of APP but not at their respective sites internal to Aβ. The identification of distinct BACE1 and BACE2 specificities and a key active-site residue important for β-site cleavage may suggest strategies for selectively inhibiting β-secretase activity. The data presented here provide insight into the mechanism of at least one form of familial AD, and suggest that BACE2 cleavage of wild-type APP within the Aβ region can limit production of intact Aβ in BACE2-expressing tissues.
Materials and Methods
Expresser Plasmid Construction and Mutagenesis.
Full-length cDNAs for APP, BACE1, and BACE2 were obtained from a U87 human glioblastoma cell (ATCC HTB14) cDNA library by PCR. Each of these constructs was ligated into a pcDNA3.1 (Invitrogen) vector modified to encode a 10-aa C-terminal tag (GTETSQVAPA) derived from bovine rhodopsin and recognized by the antibody 1D4 (21). DNA encoding the C-terminal 99 and 89 aa of APP initiated with a CD5 signal sequence was cloned into the same vector. Point mutations in APP, BACE1, and BACE2 were generated by the QuikChange (Stratagene) method.
Immunoprecipitation.
HEK293T (ATCC CRL 11554) cells (0.5 × 106) were transfected by the calcium phosphate method. Unless otherwise indicated, 4 μg of APP expresser plasmid with 0.05–0.1 μg of BACE1 or BACE2 expresser plasmids were used per transfection. Cells were metabolically labeled 24 h after transfection with 0.15 mCi of Tran35S-label (ICN) for 12 h. Harvested cells were lysed in 0.3 ml of 0.5% NP-40/0.5 M NaCl/phosphate buffer, pH 7.4, containing protease inhibitor mixture (Sigma) and 0.5 μM PMSF, and incubated for 2 h at room temperature with 1D4 antibody covalently linked to CNBr-activated Sepharose beads (Amersham Pharmacia). Washed samples were boiled in reducing sample buffer and analyzed by electrophoresis on an SDS/12% or 18% polyacrylamide gel. Immunoprecipitation of Aβ was performed by incubating tranfected cell supernatants with anti-Aβ(1–16) antibody (Chemicon) and Protein A-Sepharose beads.
Tissue Distribution of BACE2.
Multiple tissue Northern blots (Human 12-Lane, Human Brain II, and Human Brain IV) from CLONTECH were hybridized with a 3′ fragment of the BACE2-coding region (nucleotides 1120–1650). DNA was labeled with [32P]dCTP by using Ready-To-Go (Amersham Pharmacia) random oligomer-labeling beads.
Sample Preparation and Time-of-Flight Matrix-Assisted Laser Desorption/Ionization (TOF-MALDI) Mass Spectroscopy.
HEK293T cells (2 × 106) were transfected with 25 μg of BACE1 or BACE2 expresser plasmids or pcDNA3.1 alone. At 48 h after transfection, cells were lysed and immunoprecipitated as described above. Samples were washed four times with lysis buffer and twice with incubation buffer (2.5% acetic acid/ammonium acetate, pH 5.5). Immunoprecipitates bound to 1D4-Sepharose beads were mixed with 100 μl of incubation buffer containing 50 μM synthetic peptides, and incubated at 37°C for 2 h. Sepharose beads were removed by centrifugation and the 1 μl of supernatant was mixed with 1 μl of α-cyano-4-hydroxycinnamic acid matrix (Aldrich) and analyzed by TOF-MALDI mass spectroscopy (Voyager DE-STR, PerSeptive Biosystems, Framingham, MA). Laser settings of 2350 were used in all experiments. Aβ(1–28) peptide was obtained from Bachem, and the APP(662–679) peptide (KTEEISEVKMDAEFRHDS) was synthesized by New England Peptide (Lowell, MA).
Determination of pH Optima.
HEK293T cells (2 × 106) were transfected by the calcium phosphate method with 25 μg of the Swedish mutation of APP (APPsw) expresser plasmid, radiolabeled with [35S]cysteine and [35S]methionine, and lysed in 1.5 ml of unbuffered 0.5% NP-40/0.5 M NaCl containing a protease inhibitor mixture (Sigma) and PMSF. Separately, 0.4 × 106 HEK293T cells were transfected with 5 μg of plasmids encoding BACE1, BACE2, or with vector (pcDNA3.1) alone, radiolabeled, and lysed in 0.3 ml of the same lysis solution. Cell debris was removed by centrifugation. APP lysate (100 μl) was incubated on ice with 30 μl of 1.2% acetic acid/ammonium hydroxide solution prepared at indicated pH, and mixed with 5 μl of BACE1 lysate or 10 μl of BACE2 lysate and incubated at 25°C for 10 min. Ammonium hydroxide and SDS were added to achieve a final pH of 8.5. Samples were boiled, immunoprecipitated, and analyzed by SDS/PAGE.
Results
BACE2 Generates Two C-Terminal Fragments of APP.
We first investigated the ability of BACE2 to cleave APP. To do so, we generated plasmids encoding BACE1, BACE2, APP, or the C-terminal 99 or 89 aa of APP (C99 or C89, respectively). In each case, these constructs were designed to express a C-terminal tag derived from the C-terminal 9 aa of bovine rhodopsin, recognized by Sepharose beads conjugated to the antibody 1D4 (21). HEK293T cells were transfected with plasmids expressing BACE2 alone, APP alone, or APP in combination with BACE1 or BACE2. Cells were lysed, immunoprecipitated with 1D4-Sepharose, and analyzed by SDS/PAGE. Fig. 1A (lanes 3 and 4) shows that BACE2 migrates more quickly than BACE1, presumably because of the two additional N-glycosylation sites present in BACE1 (15, 20). Cells transfected with APP alone produced a C-terminal fragment which migrated slightly more rapidly than tagged C89 (Fig. 1B, lanes 2 and 5), presumably C83, a product of the endogenous α-secretase activity of these cells (22, 23). As shown in lane 3 of Fig. 1 A and B, lysates of HEK293T cells transfected with C-terminally tagged APP and BACE2 contained precipitable C-terminal fragments of APP not found in cells transfected with BACE2 or APP alone. These cells expressed a sharp band which ran identically with tagged C99 and a denser, more diffuse band which migrated more rapidly than tagged C89, and slightly more rapidly than the C-terminal fragment (C83) of cells transfected with APP alone (22). An additional diffuse band, which migrated slower than C99, was sometimes observed in the presence (Fig. 1A, lane 3) and the absence (Fig. 5A, lane 3) of full-length APP, indicating that it is not an APP cleavage product. As reported (15), cells transfected with APP and BACE1 produced two APP fragments, which comigrated with C99 and C89, respectively (Fig. 1 A and B, lane 4). We conclude that BACE2 can cleave APP at or near the β site of APP and also within the Aβ region of APP to produce a fragment smaller than the C83 product generated by the α-secretase endogenous to HEK293T cells.
Figure 1.
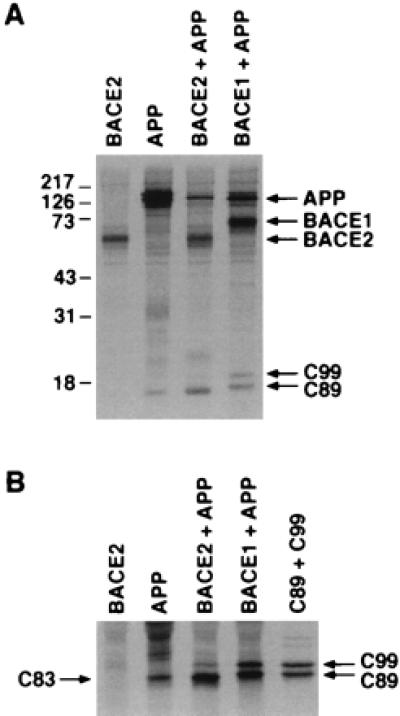
BACE2 generates two C-terminal fragments of APP. HEK293T cells were transfected with a plasmid encoding C-terminally tagged versions of the indicated molecules. Cells were radiolabeled with [35S]cysteine and [35S]methionine, lysed, and precipitated with 1D4-Sepharose beads. Precipitates were analyzed by SDS/PAGE in 12% (A) or 20% (B) polyacrylamide. In B, constructs encoding the C-terminal 99 and 89 aa of APP fused to the same C-terminal tag were included in the final lane.
Figure 5.
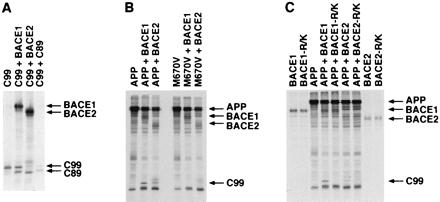
BACE1 and BACE2 β site and internal Aβ cleavage events are independent but can occur successively. (A) HEK293T cells were transfected with tagged C99 with vector alone or plasmids encoding BACE1 or BACE2 as indicated. Cell lysates were immunoprecipitated as in Fig. 1 and analyzed by SDS/PAGE. (B) HEK293T cells were transfected with wild-type tagged APP or tagged APP bearing a M671V mutation with vector alone, BACE1 or BACE2, and analyzed as in Fig. 1. (C) HEK293T were cells transfected with wild-type tagged APP alone, wild-type tagged BACE1 or BACE2, or BACE1 and BACE2 bearing an Arg → Lys mutation adjacent to their respective active sites, as indicated, and analyzed as in Fig. 1.
BACE2 Cleaves at the β Site of an APP-Derived Peptide.
To determine the preferred cleavage site of BACE2, lysates of cells transfected with plasmids encoding tagged BACE1, BACE2, or vector alone were incubated with 1D4-Sepharose beads. Beads were washed extensively and incubated with a peptide (KTEEISEVKMDAEFRHDS) derived from residues 662–679 of APP, which includes the β site (between residues M671 and D672). This peptide was then analyzed by TOF-MALDI. As shown in Fig. 2, peptide incubated with control beads exhibited a MALDI peak of 2151.0, corresponding to the full-length peptide. Fig. 2 demonstrates that the same peptide incubated with BACE1 has been cleaved to produce a strong peak at 976.4 and a smaller peak at 1193.6. The 976.4 peak corresponds to a peptide fragment (DAEFRHDS) corresponding to the first eight residues of Aβ [APP(672–679)]. The 1193.6 peak corresponds to a fragment containing the 10 aa (KTEEISEVKM) immediately upstream of the β site [APP(662–671)]. Fragments of identical molecular weight were also obtained from peptide incubated with BACE2 (Fig. 2). These data demonstrate that BACE2 can cleave the β site of an APP-derived peptide and imply that one of the C-terminal fragments observed in BACE2- and APP-transfected cells is C99.
Figure 2.

BACE2 cleaves APP(662–679) at the β site. HEK293T cells were transfected with vector alone, C-terminally tagged BACE1, or C-terminally tagged BACE2 as indicated, lysed, and precipitated with 1D4-Sepharose beads. Beads were washed and incubated with 50 μM APP(662–671) peptide and analyzed by TOF-MALDI. ++, Double protonation of the indicated peptide fragment; and *, N-terminally truncated peptide fragments observed in untreated peptide. Peaks at 1599.6 Da (BACE1) and 1606.5 and 1840.1 Da (BACE2) are not assignable to any peptide fragment. Peptide fragments of 976.4 and 1193.6 correspond to N- and C-terminal fragments of the peptide cleaved at the β site. The peptide sequence with APP and Aβ numbering is displayed below the figures.
BACE2 Cleaves Aβ(1–28) After Phe-19 and Phe-20.
We also sought to determine the site where BACE2 cleaves APP within the Aβ region of APP by using the same assay described for Fig. 2 except that a peptide derived from the first 28 aa of Aβ [Aβ(1–28)] was used. As shown in Fig. 3, peptide incubated with precipitates of mock-transfected cells was not cleaved, and a strong peak of 3261.5 corresponding to Aβ(1–28) was observed. The same peptide incubated with BACE1 produced a strong peak corresponding to the Aβ(1–10) and a peak corresponding to Aβ(11–28) consistent with the observed internal Aβ cleavage product after Tyr-10 (15). Two additional peaks corresponding to Aβ(1–19) and Aβ(1–20) were also observed, although they were substantially smaller than the Aβ(1–10) peak. When Aβ(1–28) was incubated with BACE2, pronounced peaks corresponding to Aβ(1–19) and Aβ(1–20) were observed, as was Aβ(21–28), the C-terminal counterpart of Aβ(1–20). These data are consistent with the C-terminal APP fragments observed in BACE2-transfected cells (Fig. 1) and imply that BACE2 primarily cleaves APP after Phe-19 and Phe-20 of the Aβ region to generate a product of 79–80 residues (referred to hereafter as C79).
Figure 3.

BACE2 cleaves Aβ(1–28) following Phe-19 and Phe-20. HEK293T cells were transfected with vector alone, C-terminally tagged BACE1, or C-terminally tagged BACE2 as indicated, lysed, and precipitated with 1D4-Sepharose beads. Beads were washed and incubated with 50 μM Aβ(1–28) peptide and analyzed by TOF-MALDI. ++, Double protonation of the indicated peptide. Peaks at 1196.5, 2314.2, and 2461.2 Da correspond to N-terminal peptide fragments cleaved after Aβ residues 10, 19, and 20, respectively. Peaks at 2083.6 and 966.5 Da correspond to the C-terminal peptide fragments cleaved before residues 11 and 20, respectively. The peptide sequence with APP and Aβ numbering is displayed below the figures.
The Swedish and Flemish Missense Mutations of APP Enhance Production of C99 by BACE2.
We next investigated the effect of three APP mutations implicated in early-onset familial AD on BACE1 and BACE2 proteolysis of APP. Fig. 4A demonstrates that the Swedish mutation of APP substantially increases the production of C99 by BACE2, and as previously reported, by BACE1 (14–16).
Figure 4.
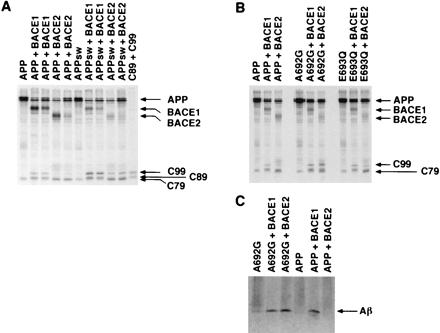
The Swedish and Flemish mutations of APP enhance the β-site cleavage product of BACE2. (A) HEK293T cells transfected with plasmids encoding wild-type tagged APP or tagged APP with the Swedish mutation (K670N/M671L; Swedish mutation of APP) with vector alone, or plasmids encoding tagged BACE1 or BACE2 as indicated. Different levels of BACE1 or BACE2 plasmid were transfected in adjacent identically labeled lanes. Cell lysates were analyzed as in Fig. 1. (B) HEK293T cells transfected wild-type tagged APP or tagged APP with the Flemish (A692G) or the Dutch (E693Q) mutations with vector alone or plasmids encoding BACE1 or BACE2 as indicated. Cell lysates were analyzed as in Fig. 1. (C) Supernatants from radiolabeled HEK293T cells transfected with plasmids encoding Flemish APP (A692G) or wild-type APP together with vector alone or plasmids encoding BACE1 or BACE2 were immunoprecipitated with an anti-Aβ(1–16) antibody and analyzed by SDS/PAGE.
Two additional mutations implicated in familial AD were assayed for their ability to alter BACE1 and BACE2 processing of APP. These mutations (A692G, the Flemish mutation, and E693Q, the Dutch mutation) are adjacent to the preferred cleavage sites of BACE2. As shown in Fig. 4B, no detectable differences in C-terminal fragment production between wild-type APP and APP with the Dutch mutation were observed when coexpressed with either BACE1 or BACE2. Contrary to a previous suggestion (8), the Flemish mutation did not significantly reduce the production of C83 (Fig. 4B, lanes 1 and 4). However, this mutation somewhat enhanced C99 and C89 production by BACE1 (lanes 2 and 5) and substantially enhanced C99 production by BACE2 to levels at or greater than those produced by BACE1 (lanes 3 and 6). Also, the Flemish mutation resulted in an additional BACE2 C-terminal fragment that migrated similarly to C89 (lane 6), consistent with the enhancement of the p3.5 fragment (Aβ starting at E11) observed in an earlier study (24). Somewhat lower levels of the BACE2-generated C79 were observed in cells transfected with BACE2 and Flemish APP (lane 6). The pronounced effect of the Flemish mutation on the level of C99 generated by BACE2 suggests a role for BACE2 in the Aβ deposition found in individuals bearing this mutation.
To determine if the Flemish mutation also increased the production of Aβ by BACE2, radiolabeled supernatants of cells transfected with wild-type APP, or APP bearing the Flemish mutation with or without BACE1 or BACE2, were immunoprecipitated with an antibody recognizing the first 16 residues of Aβ. As shown in Fig. 4C, Aβ was detectable in supernatants of cells transfected with BACE1 and Flemish or wild-type APP (lanes 2 and 5). A similar amount of Aβ was detectable in cells transfected with BACE2 and APP bearing the Flemish mutation (lane 3). Little or no Aβ was detectable in supernatants of cells tranfected with BACE2 and wild-type APP. We conclude that the Flemish mutation substantially increases the generation of Aβ as well as C99.
BACE2 Is Expressed in Neural Tissue and the Periphery.
Both BACE1 and BACE2 were cloned for this study from a cDNA library of the astroglioma cell line U87. We sought to further describe the expression patterns of BACE2. We found that BACE2 is expressed at low levels in neural tissue relative to its marked expression in the heart, kidney, and placenta (data not shown), in contrast to the expression pattern reported for BACE1, which expresses more strongly in whole brain than in these peripheral organs (15, 16). An examination of specific expression in different brain regions demonstrated that the BACE2 message was detectable at low levels in all such areas, with the exception of the spinal cord and medulla where it was efficiently expressed (data not shown). These data indicate that BACE2 is expressed in regions of the brain also expressing BACE1 (15, 16) and suggest that BACE2 may play a role in highly vascularized systemic tissues, including the heart, kidney, and placenta.
BACE1 and BACE2 Can Cleave C99.
We next investigated whether BACE1 or BACE2 could cleave C99 and thus continue to process an APP fragment already cleaved at its β site. Fig. 5A demonstrates that both BACE1 and BACE2 can cleave a tagged C99 construct, producing fragments that run at (BACE1, lane 2) or below (BACE2, lane 3) tagged C89, respectively. No endogenous α-secretase cleavage of C99 could be detected in this experiment (lane 1). The efficiency with which each enzyme cleaves C99 closely matches the efficiency with which they cleave full-length APP at their respective cleavage sites internal to Aβ. These data indicate that both enzymes can continue to process C-terminal APP fragments after initial β-site cleavage of APP.
An M671V Mutation in APP Inhibits Both BACE1 and BACE2 β-Site Cleavage.
We next assayed the ability of a M671V mutation immediately upstream of the β site to interfere with BACE1 and BACE2 proteolysis. M671V has been shown to inhibit the production of Aβ in HEK293 cells transfected with APP bearing this mutation (25). Fig. 5B demonstrates that this mutation almost completely inhibited both BACE1 and BACE2 cleavage at the β site of APP but did not significantly alter the generation of their respective internal cleavage products. These data indicate that the β site and the internal cleavage sites of BACE1 and BACE2 can be cleaved independently and underscore the similarity of the active sites of these two enzymes.
A Key Active Site Arg Is Important for β-Site Cleavage by BACE1 and BACE2.
Inspection of the crystal structure of the aspartyl protease cathepsin D bound to its inhibitor pepstatin (26) and alignment of cathepsin D, BACE1, and BACE2 sequences suggested that Arg-296 of BACE1 (Arg-310 of BACE2) could form a salt bridge with the Asp immediately after the β site of APP (APP residue 672, Aβ residue 1). We speculated that the internal Aβ proteolysis by BACE2 would be less sensitive to perturbations of this Arg because these sites did not include an immediately adjacent acidic amino acid (shown in Fig. 6B). We also speculated that the longer Glu found immediately C-terminal to the internal BACE1 cleavage site (Fig. 6B) would be better able to associate with a shorter Lys in the enzyme. Fig. 5C demonstrates that an Arg-296 → Lys of BACE1, and the analogous change at position 310 of BACE2, do indeed interfere with the ability of both enzymes to cleave at the β site of APP, but not at their respective cleavage sites internal to Aβ. These data are consistent with the formation of a salt bridge between Asp-672 of APP (Aβ residue 1) and Arg-296 and Arg-310 of BACE1 and BACE2, respectively.
Figure 6.

Preferred BACE1 and BACE2 cleavage sites. (A) Sequence of APP indicating α- and β-cleavage sites, BACE1- and BACE2-cleavage sites, and the location of mutations analyzed here. APP numbering is that of the 770-aa isoform. (B) Alignment of BACE1 and BACE2 substrates with the relative efficiency of cleavage indicated by pluses. Residues that are suggested here to modulate the efficiency or specificity of proteolysis by these enzymes are indicated in bold. Asterisks indicate that although little C-terminal fragment derived from BACE1 cleavage at these sites was observed in HEK293T cells, BACE1 could cleave Aβ(1–28) at these sites in an in vitro assay (Fig. 3).
Maximal BACE2 Activity Occurs at Acidic pH.
β-Secretase activity occurs with maximum efficiency under acidic conditions (15). To determine the pH profile of BACE2, lysates from radiolabeled HEK293T cells transfected with APP containing the Swedish mutation were mixed with lysate of radiolabeled cells transfected with BACE1 or BACE2 and incubated in buffer ranging in pH from 3 to 7.5. As reported (15), BACE1 has an acidic pH optimum which we observed to occur at ≈ pH 5.0 (data not shown). BACE2 exhibited a similar pH profile, with its maximal activity occurring close to pH 5.5 (data not shown). These data further underscore the close functional similarity between these two enzymes.
Discussion
Aβ is the initial and major component of amyloid plaques observed in the brains of all individuals with AD, and mutations in the APP or presenilin proteins implicated in familial AD alter either the quantities or the forms of Aβ produced (2). BACE1 likely makes a significant contribution to the pool of Aβ present in the brain, and therefore this protease is likely to be an important therapeutic target in AD treatment (13–16).
Here we characterize a homologous enzyme, BACE2, which we show is also a BACE. Like BACE1, BACE2 efficiently cleaves sites internal to the Aβ region of APP. Although both enzymes cleave within Aβ, the fragments of Aβ produced by these internal cleavages may have different clinical consequences. BACE1-generated Aβ fragments beginning at Glu-11 of Aβ have been observed in senile plaques (18), and fragments of this size have been shown to be more amyloidogenic and more neurotoxic than full-length Aβ (27). It may also be important that the BACE1-generated Aβ fragments, like full-length Aβ, include the HHQK sulfate-binding region of Aβ, which can associate with sulfated proteoglycans found in senile plaques (28, 29). In contrast, BACE2-cleaved internal fragments (starting at Aβ Phe-19 and Phe-20) lack the HHQK domain and have not to date been observed in senile plaques. Moreover, fragments of the size of p3 (starting at Aβ Leu-17) or smaller appear to be less amyloidogenic and neurotoxic in tissue culture (27). We have shown that BACE2 is more efficient at cleaving within Aβ than BACE1 and less efficient at generating C99 (Fig. 1). We have also demonstrated BACE2 can efficiently degrade C99 (Fig. 5A). Taken together, these observations imply that BACE2 might limit the production of pathogenic forms of Aβ (i.e., fragments beginning at Asp-1 or Glu-11) in cells that express both BACE1 and BACE2.
The Flemish mutation of APP significantly alters the ratio of BACE2-generated C99 to its arguably more benign internal cleavage product (C79) and also gives rise to an additional BACE2 product which migrates similarly to C89 (Fig. 4B), in agreement with the results of Haass et al. (24). The mechanisms underlying this pronounced increase in C99 remain unclear. It has previously been suggested that the Flemish mutation interferes with constitutive cleavage by an α-secretase (8), but our data show no significant effect of this mutation on endogenous α-secretase cleavage of APP. We do, however, detect a decrease in the generation of the internal BACE2 cleavage product (C79). Perhaps the Flemish mutation at Aβ residue 21 inhibits the BACE2 cleavage after residues 19 or 20, thereby blocking the further proteolysis of BACE2-generated C99.
The tissue distribution of BACE2 is also consistent with a role for BACE2 in the particular form of AD found in patients bearing the Flemish mutation. Although BACE2 is expressed at relatively low levels in the cortex, a shift in its activity from limiting to producing amyloidogenic Aβ fragments (i.e., starting at Asp-1) could contribute to the large senile plaques observed in these patients (11). High expression of BACE2 in the kidney, placenta, and heart is consistent with the presence of BACE2 in the vasculature, and a role for BACE2 in the angiopathy and cerebral hemorrhages observed in the Flemish patients (8).
The increased β-site proteolysis of Swedish mutation of APP by BACE2 further underscores the similarity in the active sites of BACE1 and BACE2, as does the ability of the conservative M671V mutation to ablate the β-site cleavage of both enzymes (Fig. 5B). The similar response to both β-site mutations implies that most active-site inhibitors of BACE1 will also inhibit BACE2. Our data raise the possibility that it may be advantageous to inhibit BACE1 specifically, both because BACE2 may contribute to the degradation of Aβ in the brain and because it may have other significant roles in the periphery. Alignment of the preferred cleavage sites of BACE1 and BACE2 (Fig. 6B) suggests that one difference between them is the position of acidic amino acids. In particular, an acidic amino acid at the third position after the cleavage site may be important for efficient BACE2 activity. Similarly, acidic residues at the fourth position preceding and at the first position after the cleavage site may play a role in BACE1 cleavage. These possibilities still need to be explored. However, it is already clear from our data that BACE1 but not BACE2 cleaves Aβ after Tyr-10 (Fig. 3). Peptide variants based on the sequence of this site could presumably serve as models for small molecules that specifically inhibit BACE1.
Most AD is not clearly familial, and its etiology has not been defined, although a role for excessive Aβ deposition is clear. Evidence presented here suggests that BACE2 and BACE1 function together to regulate the production of Aβ peptides. It may therefore be useful to explore the consequences of the natural variation in the activities of BACE2 as well as BACE1.
Acknowledgments
We thank Joseph Sodroski, Norma Gerard, and Craig Gerard for their guidance, Dennis Selkoe for his thoughtful comments and careful reading of the manuscript, Miriam Ritz for her assistance, and Minou Modabber for her excellent assistance with graphics.
Abbreviations
- Aβ
amyloid-β protein
- APP
Aβ precursor protein
- β site
N terminus of Aβ
- BACE
β-site APP-cleaving enzyme
- AD
Alzheimer's disease
- C99
membrane-associated C-terminal APP fragment of 99 aa
- C83
C-terminal APP fragment of 83 aa
- C89
C-terminal APP fragment of 89 aa
- TOF-MALDI
time-of-flight matrix-assisted laser desorption/ ionization
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160115697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160115697
References
- 1.Price D L, Sisodia S S. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe D J. Nature (London) 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 3.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung A Y, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe D J. Nature (London) 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 4.Goate A, Chartier-Harlin M C, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Nature (London) 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 5.Chartier-Harlin M C, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. Nature (London) 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 6.Murrell J, Farlow M, Ghetti B, Benson M D. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki N, Cheung T T, Cai X D, Odaka A, Otvos L, Jr, Eckman C, Golde T E, Younkin S G. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 8.Hendriks L, van Duijn C M, Cras P, Cruts M, Van Hul W, van Harskamp F, Warren A, McInnis M G, Antonarakis S E, Martin J J, et al. Nat Genet. 1992;1:218–221. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 9.Levy E, Carman M D, Fernandez-Madrid I J, Power M D, Lieberburg I, van Duinen S G, Bots G T, Luyendijk W, Frangione B. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 10.Van Broeckhoven C, Haan J, Bakker E, Hardy J A, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos R A. Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 11.Cras P, van Harskamp F, Hendriks L, Ceuterick C, van Duijn C M, Stefanko S Z, Hofman A, Kros J M, Van Broeckhoven C, Martin J J. Acta Neuropathol. 1998;96:253–260. doi: 10.1007/s004010050892. [DOI] [PubMed] [Google Scholar]
- 12.Bornebroek M, Haan J, Maat-Schieman M L, van Duijnen S G, Roos R. Brain Pathol. 1996;6:111–114. doi: 10.1111/j.1750-3639.1996.tb00793.x. [DOI] [PubMed] [Google Scholar]
- 13.Hussain I, Powell D, Howlett D R, Tew D G, Meek T D, Chapman C, Gloger I S, Murphy K E, Southan C D, Ryan D M, et al. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 14.Sinha S, Anderson J P, Barbour R, Basi G S, Caccavello R, Davis D, Doan M, Dovey H F, Frigon N, Hong J, et al. Nature (London) 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 15.Vassar R, Bennett B D, Babu-Khan S, Kahn S, Mendiaz E A, Denis P, Teplow D B, Ross S, Amarante P, Loeloff R, et al. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 16.Yan R, Bienkowski M J, Shuck M E, Miao H, Tory M C, Pauley A M, Brashier J R, Stratman N C, Mathews W R, Buhl A E, et al. Nature (London) 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 17.Haass C, Schlossmacher M G, Hung A Y, Vigo-Pelfrey C, Mellon A, Ostaszewski B L, Lieberburg I, Koo E H, Schenk D, Teplow D B, et al. Nature (London) 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 18.Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses A D, Tjernberg L O, Silberring J, Gandy S E, Winblad B, Greengard P, et al. Proc Natl Acad Sci USA. 1994;91:8378–8382. doi: 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al. Nature (London) 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 20.Saunders A J, Kim T, Tanzi R E. Science. 1999;286:1255a. (technical comment). [Google Scholar]
- 21.Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard N P, Gerard C, Sodroski J, Choe H. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 22.Esch F S, Keim P S, Beattie E C, Blacher R W, Culwell A R, Oltersdorf T, McClure D, Ward P J. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Meschia J F, Cotter R J, Sisodia S S. J Biol Chem. 1991;266:16960–16964. [PubMed] [Google Scholar]
- 24.Haass C, Hung A Y, Selkoe D J, Teplow D B. J Biol Chem. 1994;269:17741–17748. [PubMed] [Google Scholar]
- 25.Citron M, Teplow D B, Selkoe D J. Neuron. 1995;14:661–670. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin E T, Bhat N, Gulnik S, Hosur M V, Sowder R C, Cachau R E, Collins J, Silva A M, Erickson J W. Proc Natl Acad Sci USA. 1993;90:6796–6800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pike C J, Overman M J, Cotman C W. J Biol Chem. 1995;270:23895–23898. doi: 10.1074/jbc.270.41.23895. [DOI] [PubMed] [Google Scholar]
- 28.Giulian D, Haverkamp L J, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher A E. J Biol Chem. 1998;273:29719–29726. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 29.Snow A D, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wright T N. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]