

Abstract
Neocortical pyramidal neurons have extensive axonal arborizations that make thousands of synapses. Action potentials can invade these arbors and cause calcium influx that is required for neurotransmitter release and excitation of postsynaptic targets. Thus, the regulation of action potential invasion in axonal branches might shape the spread of excitation in cortical neural networks. To measure the reliability and extent of action potential invasion into axonal arbors, we have used two-photon excitation laser scanning microscopy to directly image action-potential-mediated calcium influx in single varicosities of layer 2/3 pyramidal neurons in acute brain slices. Our data show that single action potentials or bursts of action potentials reliably invade axonal arbors over a range of developmental ages (postnatal 10–24 days) and temperatures (24°C-30°C). Hyperpolarizing current steps preceding action potential initiation, protocols that had previously been observed to produce failures of action potential propagation in cultured preparations, were ineffective in modulating the spread of action potentials in acute slices. Our data show that action potentials reliably invade the axonal arbors of neocortical pyramidal neurons. Failures in synaptic transmission must therefore originate downstream of action potential invasion. We also explored the function of modulators that inhibit presynaptic calcium influx. Consistent with previous studies, we find that adenosine reduces action-potential-mediated calcium influx in presynaptic terminals. This reduction was observed in all terminals tested, suggesting that some modulatory systems are expressed homogeneously in most terminals of the same neuron.
The complex axonal arbors of neocortical pyramidal neurons are a defining feature of cortical circuits (1). These axons form en passant presynaptic terminals that appear as small (diameter ≈1 μm) swellings of the axon (2). When action potentials invade axonal arbors, calcium enters presynaptic terminals through voltage-sensitive calcium channels (VSCCs) causing release of neurotransmitter (3, 4). Do action potentials reliably invade each branch of the axonal arbor? The answer to this question has profound implications for the spread of excitation through neural networks (5). For example, modeling studies have suggested that action potentials could fail at axonal branch points in an activity-dependent manner (6). Depending on the temporal pattern of action potentials, such mechanisms can act in some systems to effectively silence groups of synapses on parts of the axonal arbor that are downstream of the point of failure (7, 8). Because of their small size (1), most axons and presynaptic terminals have remained out of reach for direct electrophysiological measurements. Modern imaging techniques provide an alternative approach to study excitation in these structures. In particular, microspectrometric measurements of intracellular free calcium concentration, [Ca2+], has become an important tool to characterize presynaptic action potential invasion and the dynamics of the resulting calcium current (9, 10) and accumulation (11, 12). However, measurements of presynaptic [Ca2+] have so far been mostly limited to populations of presynaptic terminals (9, 10), specialized large synaptic terminals (11, 13, 14), or proximal axons (15). If action-potential-evoked [Ca2+] transients could be resolved in single cortical terminals, imaging would provide a powerful tool to study action potential invasion into individual branches of the complex cortical axonal arbors. In addition, measurement of presynaptic [Ca2+] in single terminals could shed light on possible mechanisms underlying fluctuations in neurotransmitter release. Several previous studies employing [Ca2+] imaging of individual central nervous system terminals have produced conflicting results. In cultured cortical neurons, action potentials appear to reliably invade the entire axonal arbor, producing [Ca2+] transients with small trial-to-trial variability (16, 17). In contrast, imaging experiments in neocortical brain slices from young animals (11–16 days old) showed large trial-to-trial variability, including failures of [Ca2+] increases (18). In cerebellar basket cell axons, calcium influx evoked by single action potentials appears difficult to detect (19). Action potential propagation into cortical axonal arbors has also been studied by using more indirect electrophysiological approaches. For example, experiments employing minimal stimulation in hippocampal slices have provided support for reliable action potential propagation (20). Recently, recordings from pairs of connected neurons in cultured hippocampal brain slices have suggested the interesting possibility that action potential propagation may be controlled by an IA-like K+ current in the proximal axon in a branch-specific manner (21). This effect would provide a powerful mechanism to control the flow of excitation in axonal arbors by the recent activity of the neuron.
In this study we have directly investigated action potential propagation into branches of axonal arbors of layer 2/3 neocortical pyramidal cells, which make synapses with a wide range of postsynaptic targets. We have taken advantage of two-photon excitation laser scanning microscopy (2PLSM; refs. 22 and 23) to observe [Ca2+] transients in single varicosities in response to somatically evoked action potentials in intact neural circuits of the brain slice. Electronmicroscopic analysis has revealed that virtually all axonal varicosities of pyramidal neurons contain synaptic terminals (24, 25). [Ca2+] elevations in the axonal varicosities evoked by action potentials are indicative of local action potential invasion. We can, therefore, directly observe depolarization produced by action potentials in regions of the axon that are electrotonically distant from the soma. Our data show that, in layer 2/3 pyramidal cells, action potentials radiate reliably into axons under a large set of experimental conditions.
Methods
Rat neocortical slices were prepared by using published procedures (26). Sprague–Dawley rats (P10-P24) were deeply anesthetized with pentobarbital sodium (55 mg/kg) and decapitated, and the brains were quickly removed and placed in cold, oxygenated slicing solution containing (in mM): 2.5 KCl, 1.25 NaH2PO4, 7.0 MgCl2, 0.5 CaCl2, 25.0 NaHCO3, 25.0 glucose, 110.0 choline chloride, 11.6 ascorbic acid, and 3.1 pyruvic acid. Neocortical slices (350–450 μm) were cut in the coronal plane with a vibratome and then placed in a holding chamber (34°C) for ≈1 h before recording. Individual slices were transferred to a submersion-type recording chamber continuously perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 126.0 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 MgCl2, 2.0 CaCl2, 26.0 NaHCO3, and 10.0 glucose (pH 7.4).
Whole-cell recordings were obtained from layer 2/3 pyramidal neurons of sensorimotor cortex under visual control (n = 47). The depth of recorded neurons in the slice was in the range 40–80 μm. The recording pipettes were filled with intracellular solution containing (in mM): 130.0 potassium methylsulfate, 4.0 MgCl2, 4 Na2-ATP, 0.4 Na-GTP, 10.0 Hepes, 10 Na-phosphocreatine, and Oregon green 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA) I (OGB1; 100–300 μM). In intracellular solution, OGB1 has a high affinity for calcium, Kd ≈206 nM (27). Approximately half of the experiments were performed at 30°C (n = 24), and the remaining experiments were performed at 24°C–26°C (n = 23). Single or multiple action potentials were evoked by passing short (10–150 ms) depolarizing pulses (20–200 pA, current clamp; 20–40 mV, voltage clamp) through the recording pipette. Control experiments showed that depolarizations that did not evoke action potentials at the soma did not produce calcium concentration transients in the axon. Fluorescence was measured in single axon varicosities by using 2PLSM (22, 23). Our custom microscope was based on a Ti:sapphire laser (Mira 900F; Coherent, Santa Clara, CA) running at 800 nm and a high n.a. water immersion objective (×63, n.a. = 0.9; Zeiss), with software written at Bell Laboratories (Ray Stepnoski). Fluorescence signals were collected through both objective and condenser and summed (28). Data were collected by using frame scans or line scans (28). Frame scans consisted of a series of 64 × 64 pixel images (128 ms/image) collected before and after evoking action potentials in the neuron. Although frame scans are relatively slow (128 ms per frame), they inform about the spatial structure of [Ca2+] dynamics. Line scans were collected by scanning the laser beam back and forth across the structure of interest in a line (2 ms/line). Line scans allow excellent time resolution (2 ms per line) at the expense of sacrificing one spatial dimension. Care was taken to limit indicator dye expelled into the extracellular space during patching, ensuring that the background because of exogenous dye was minimal. For analysis, the background was subtracted, and the change in fluorescence over the resting fluorescence, ΔF/F = (F − F0)/F, was computed; this measure is roughly proportional to changes in [Ca2+] (27). Data are shown as mean ± SD.
Results
Whole cell recordings were made from 47-layer 2/3 pyramidal neurons from animals spanning a range of developmental ages (postnatal day 10–24). Because no obvious differences were observed for different ages, the data were pooled. After break-in, we allowed 10 min before imaging to allow the calcium indicator to equilibrate with the proximal axonal arbor (distance ≈100–500 μm). Axons were recognizable in layer 2/3 pyramidal neurons as processes pointing toward the white matter (Fig. 1A). Furthermore, axons are smaller and therefore substantially dimmer than dendrites, and they lack spines. When following axons from the soma under 2PLSM, axonal branches and swellings were encountered (Fig. 1 A and B). The swellings, short bright stretches of axon, resembled en passant type axonal varicosities, and it is from these structures that the majority of our calcium measurements were obtained. Measurements were made from single varicosities on 2nd- to 4th-order axonal branches that were at least 100 μm from the soma (range 100–500 μm). In most recordings, only a single varicosity could be imaged before the signal ran down. In six neurons, multiple varicosities were imaged. ΔF/F was measured in response to action potentials initiated by depolarizing current injections via the somatic recording pipette (Fig. 1A).
Figure 1.
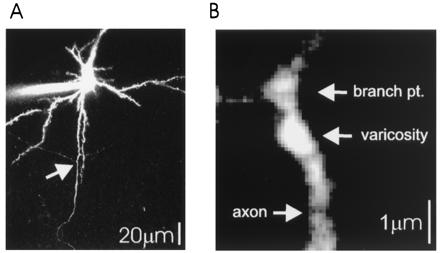
Imaging of axonal arbors of layer 2/3 pyramidal cells. (A) Low magnification image of a neuron filled with 100 μM OGB1, showing the apical dendrite (Top), several spiny basal dendrites, and the primary axon with two primary to secondary branch points (arrow). Note the low fluorescence intensity of secondary axonal branches compared with basal dendrites, consistent with the small diameters of axons. (B) High magnification image of a branch point between secondary and tertiary branches (same axon as in 2C). An axonal swelling is clearly recognizable by large resting fluorescence. Axonal branch points also show relatively large fluorescence.
Can [Ca2+] transients evoked by single action potentials be observed in individual varicosities as they can in single synaptic spines (29)? To address this question, we imaged fluorescence in single terminals by using frame and line scans while action potentials were evoked at the soma. In both imaging modes, it was possible to detect [Ca2+] transients produced by single action potentials (Fig. 2). Action potential-evoked fluorescence changes varied greatly between different varicosities. In some structures, [Ca2+] transients were difficult to detect above background (data not shown), whereas in others large fluorescence changes were observed (range of analyzed signal amplitudes, 25%–118%; mean 56% ± 25%; Fig. 2). Variations between different varicosities could be due to differences in calcium influx, surface to volume ratio, resting [Ca2+] (and hence differences in resting fluorescence), or calcium buffering. The source of the differences between different terminals was not investigated further.
Figure 2.
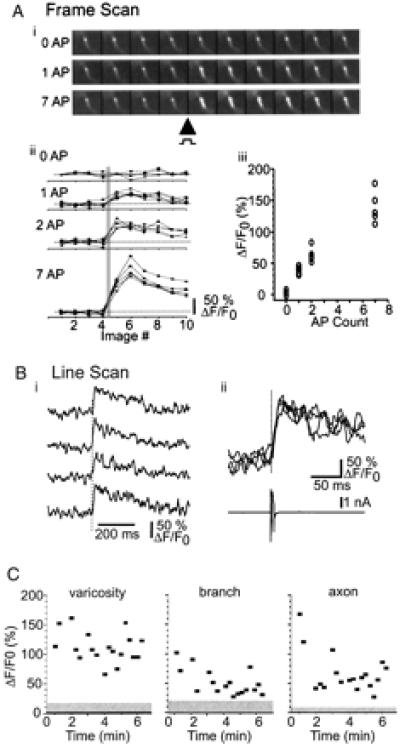
Action-potential-evoked [Ca2+] transients in individual synaptic terminals. (A) (i) Frame scans from a single varicosity on a 2nd order axonal branch. (ii) Sequential image scans for 0, 1, 2, and 7 evoked action potentials. The solid gray line indicates timing of action potentials. (iii) Relationship between action potential number and [Ca2+] transient amplitude (three to five trials each). (B) Line scans from the same varicosity as image scans in A, showing rapid rise time of [Ca2+] transients. (i) Repeated trials illustrating [Ca2+] transients in response to single action potential. (ii) Overlay of data from i with somatic electrophysiology showing action potential in voltage clamp. (C) Amplitudes of [Ca2+] transients evoked by single action potentials in varicosities (Left), branch points (Middle), and axon (Right) (same site as in Fig. 1B). Gray regions indicate the rms noise level in trials lacking action potentials. In this example, fluorescence measurements started soon after break-in. Action-potential-evoked [Ca2+] transient amplitudes decreased with time, consistent with increasing calcium buffer concentrations.
[Ca2+] transients were not restricted to axonal varicosities but were also observed in axons and in the swellings at axonal branches (Fig. 2C). However, [Ca2+] transients evoked by action potentials were typically larger in varicosities than in axons (Fig. 2C). Taken together with the fact that surface-to-volume ratios are smaller in varicosities compared with axonal shafts [factor 2–3 (25)], our data suggest that VSCCs in cortical axons are concentrated at varicosities.
Are action potential-evoked [Ca2+] transients reliable? [Ca2+] transients measured in single varicosities report on local action potential invasion. Large trial-to-trial fluctuations in [Ca2+] transients, including failures, could be caused by unreliable action potential invasion or, in case of small number of VSCCs, unreliable coupling between depolarization and opening of channels. On the other hand, small trial-to-trial fluctuations in [Ca2+] transients would be proof that action potentials invade axonal arbors reliably and would set limits on the variability of coupling between depolarization and opening of VSCCs. For this purpose, we focused our attention on varicosities that showed large fluorescence transients (Figs. 2–4). In none of the 52 varicosities that were tested in this way were failures of [Ca2+] transients in response to single action potentials observed (10 to 96 trials; mean = 35 trials/varicosity) (Figs. 2–4). Thus, varicosities that responded with clear [Ca2+] transients to action potentials continued responding without failure to every trial for the duration of the recording, suggesting that action potentials reliably invade axonal arbors.
Figure 4.
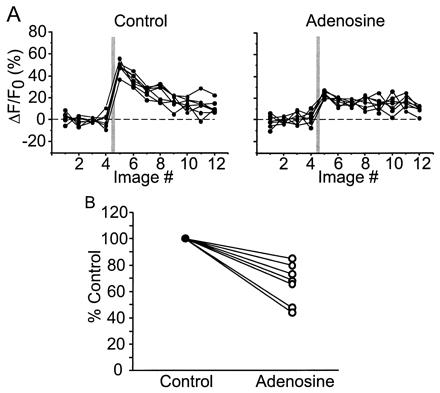
Adenosine suppresses [Ca2+] transients in single axonal varicosities. (A) Image scans from a single varicosity. In control conditions, clear [Ca2+] transients were observed in response to single action potentials. After bath application of adenosine (50 μM), [Ca2+] transients were significantly reduced. Each panel consists of seven consecutive trials in each condition. (B) Summary of adenosine effect in seven neurons.
Could action potentials fail in an activity-dependent manner? To begin testing this possibility, we evoked small trains of action potentials (2–3 spikes; interspike interval 10–20 ms). These bursts reliably produced [Ca2+] transients whose amplitudes were roughly proportional to number of action potentials, suggesting that each spike in those bursts is propagated reliably into axonal arbors (Fig. 2A). Bursts containing larger numbers of action potentials (4–10 action potentials) produced even larger [Ca2+] transients, but fluorescence increased in a sublinear fashion with action potential number (Fig. 2A). Because we used a high-affinity [Ca2+] indicator (Kd ≈200 nM), this nonlinearity probably reflects saturation of the indicator (27, 30). The dependence of [Ca2+] transient amplitudes on action potential number can be used to estimate the maximum change in fluorescence produced by saturating [Ca2+], ΔFmax/F. This quantity, which is useful for computing absolute [Ca2+] levels (27), was found to be in the range ΔFmax/F ≈200%–300%.
Recent experiments in cultured hippocampal slices have suggested that deinactivation of an IA-like K+ current by somatic hyperpolarization may inhibit action potential invasion into branches of the axonal arbor (21, 31, 32). Previous studies have reported 4-aminopyridine-sensitive currents in rat neocortical pyramidal cells (see ref. 33 and citations therein). Consistent with these studies, in our preparation, application of 4-aminopyridine (100 μM) reduced a depolarization-activated hyperpolarizing current (n = 2). Using current-clamp recordings, we tested whether such conductances are indeed activated and have an effect on action potential propagation in axons of cortical pyramidal cells. The depolarizing pulses used to evoke action potentials were preceded by hyperpolarizing somatic current injections of variable length (110–300 ms) and amplitude (200–600 pA) (Fig. 3A). In nine of nine experiments, we failed to observe differences between [Ca2+] transients evoked by action potentials alone or action potentials preceded by a variety of hyperpolarizing steps (Fig. 3). Because [Ca2+] transient amplitudes and frequencies were not modulated by stimuli that ought to deinactivate IA, it appears that action potential propagation is not controlled by IA-like K+ currents in cortical pyramidal neurons.
Figure 3.
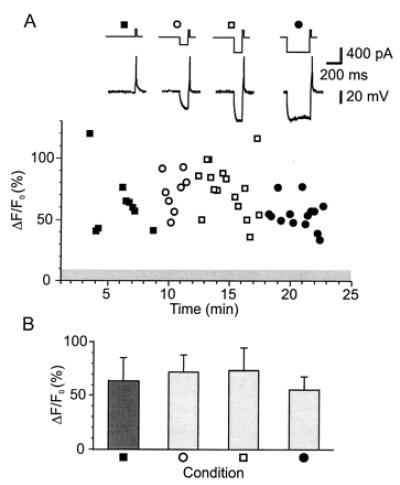
Action potentials reliably produce [Ca2+] transients in single varicosities regardless of prior membrane potential manipulations. (A, Top) Current injection protocol used to evoke single action potential in each set of trials. (Middle) Voltage response to above protocol by somatic recordings. (Bottom) Fluorescence response amplitudes. The data plotted are peak amplitudes (ΔF/F) obtained from line scan images. In control conditions, a single depolarizing pulse evoked a single action potential, producing [Ca2+] transients (closed squares). In the next series of trials, hyperpolarizing current pulses (200 pA, 110 ms), applied to activate IA, preceded the depolarizing pulse by 2 ms. These pulses had no effect on the [Ca2+] transient amplitudes (open circles). Increasing the amplitude of the hyperpolarizing current pulse to 400 pA (open squares) and then the duration to 300 ms (closed circles) produced no significant change in the [Ca2+] transients. No failures were observed under any condition. The gray bar indicates the mean ± SD of noise measures (no stimulus). (B) The data plotted are the average responses (±SD) in each condition listed above. Essentially identical results were collected for eight other varicosities.
It has been proposed that temperature may modulate the safety factor of action potential propagation in axonal arbors (6). Surprisingly, [Ca2+] transients measured at 24°C were comparable in amplitude to those measured at 30°C (53% ± 12% and 63% ± 30%, respectively). Because we focused our measurements on terminals showing measurable signals, this similarity might be an artifact of sampling. However, stochastic failures of action potential-evoked [Ca2+] transients were never observed at either temperature, suggesting that invasion was reliable over the whole range of experimental temperatures.
Activation of a variety of neurotransmitter and neuromodulator receptors at presynaptic terminals controls important aspects of synaptic transmission (12) (34). For some inhibitory modulators, for example adenosine, one mechanism underlying presynaptic inhibition is reduction of action-potential-mediated calcium influx (12) (35). However, because previous studies were based on measurements from populations of presynaptic terminals and axons, it is not clear whether these mechanisms of presynaptic inhibition exist at all terminals or are restricted to a subset of terminals. To determine how ubiquitous adenosine is as a neuromodulator, we tested whether adenosine does alter the magnitude of action-potential-evoked [Ca2+] transients at individual varicosities. After establishing a consistent baseline response (Fig. 4A, control), adenosine (50 μM) was added to the ACSF. In the presence of the drug, the [Ca2+] transients were significantly attenuated in all varicosities tested (Fig. 4A, adenosine). For the population of varicosities tested, adenosine significantly reduced the [Ca2+] transient (to 66.4% ± 15.4% of control, P < 0.01, Wilcoxon test; n = 7; Fig. 4B), without changing the decay time (control: 437 ± 182 ms; adenosine, 486 ± 188 ms). Our data suggest that presynaptic inhibition of calcium influx exists at most cortical presynaptic terminals.
Discussion
When imaging in scattering tissue, 2PLSM distinguishes itself by providing diffraction-limited resolution together with efficient detection of fluorescence (22, 23). These properties are essential for imaging [Ca2+] dynamics in small neuronal compartments such as dendritic spines (26, 29) and especially presynaptic terminals (23). Axon diameters are only on the order of ≈300 nm, smaller by a factor of 2–3 than even small dendritic branches (1) and therefore 4–9 times dimmer in fluorescence microscopy when labeled with a cytosolic dye (Fig. 1A). In this study, we used 2PLSM to image [Ca2+] transients evoked by single action potentials in individual varicosities in axonal arbors of layer 2/3 pyramidal neurons. We used this technique to determine whether action potentials reliably invade neocortical axonal arbors.
Our data show that action-potential-evoked [Ca2+] transients in varicosities have fast rise times (<2 ms, our chosen time resolution), consistent with essentially instantaneous calcium influx during the action potential (9, 10). [Ca2+] transients had long decay times (432 ± 231 ms, at 100 μM dye concentration, n = 7). These decay times reflect the presence of exogenous calcium buffer in the form of the calcium indicator. The effect of the added buffer is characterized by its buffer capacity (the ratio of dye concentration over its affinity for calcium, ≈500 for 100 μM of OGB1) (27, 36–38). [Ca2+] transient amplitudes and durations shrink and lengthen respectively with increasing buffer capacities. If we assume that presynaptic buffer capacities are comparable with measured dendritic buffer capacities in cortical neurons (≈100) (27, 38), the durations of calcium transients in the zero added buffer limit could be as much as ≈6 times faster than in the presence of the dye, and thus comparable to the decay times reported in dendrites (<100 ms) (27, 38).
The amplitudes of [Ca2+] transients varied greatly among terminals. These differences could be due to differences in calcium current densities in different compartments. However, based on our measurements, we cannot exclude the possibility that these differences are due to variations in resting [Ca2+] or local calcium buffering. Amplitudes of [Ca2+] transients were typically smaller in axons than presynaptic terminals, despite the fact that axonal surface-to-volume ratios make axons more favorable to produce relatively larger [Ca2+] transients (25). These results suggest that VSCCs must exist at substantially higher areal densities at presynaptic terminals compared with axonal shafts. Because of the small distances between neighboring terminals (≈5 μm), it is difficult to exclude the possibility that in many instances fluorescence signals observed in axonal shafts were due to diffusion of calcium bound to indicator from neighboring varicosities, leaving open the possibility that axonal shafts are entirely free of VSCCs.
The intracellular dynamic range of OGB1 in varicosities was ΔFmax/F >200%. Single wavelength fluorescence measurements can be converted to resting calcium concentrations as [Ca2+]0 = Kd(1 − Rf−1)/δfmax − KdRf−1 (27). Rf is the ratio of fluorescences of the indicator in the calcium bound over the calcium free forms [for OGB1, Rf ≈7 (27)] and δf = (ΔF/F)/100%. Therefore, resting calcium in at least some synaptic terminals is low ([Ca2+]0 < 60 nM). Similarly, changes in [Ca2+] can be estimated (27) as
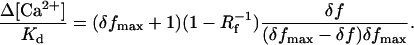 |
Because ΔF/F evoked by single action potentials was in the range 25%–120% (100 μM OGB1), [Ca2+] changes were in the range 25–240 nM (using δfmax ≈2.5). In conditions of zero added buffer, [Ca2+] transients may thus reach amplitudes in excess of 1000 nM, severalfold larger than those seen in apical dendrites (27, 38).
Repeated firing of action potentials produced [Ca2+] transients with small trial-to-trial variability (Figs. 2–4) (range of SD/mean = 0.18–0.60). In other words, if in a given varicosity an action-potential-evoked [Ca2+] transient was observed on one trial, a similar transient was observed on other trials. This result argues strongly against spontaneous fluctuations in action potential invasion into axons. It is thus unlikely that branch-point failures underlie fluctuations in neurotransmitter release observed in postsynaptic neurons (20). However, our data do not exclude the possibility that local trial-to-trial fluctuations in the action-potential-evoked calcium influx, perhaps in nanodomains close to the release apparatus (13), might underlie stochastic quantal release (18).
The magnitudes of fluorescence transients were proportional to the number of evoked action potentials for small bursts (1–3 spikes; interspike interval <20 ms). We conclude that even short bursts of action potentials are transmitted reliably throughout the axonal arbor of neocortical pyramidal neurons. For longer trains, fluorescence transients showed saturating behavior consistent with saturation of our high-affinity calcium indicator (Fig. 2A), which is still consistent with reliable action potential invasion.
Previous electrophysiological studies have suggested that manipulation of the membrane potential at the soma before action potential initiation can suppress spread of action potentials in branch-specific ways (21). This effect was attributed to a 4-aminopyridine sensitive current (IA) that is inactivated at rest, allowing action potential propagation to proceed, but deinactivates at hyperpolarized potentials, shunting action potential propagation. Thus, hyperpolarization could serve as a mechanism for branch point propagation control (21). In our recordings, strong membrane hyperpolarization, which should deinactivate IA-like currents, before evoking a single action potential, had no effect on the [Ca2+] transients in the varicosities, axonal branch points, or neighboring axon near the varicosity. These data suggest that IA does not regulate propagation of the action potential through the axonal arbor within neocortical pyramidal neurons. Differences in brain regions (hippocampus vs. neocortex) or preparation (slice culture vs. acute slice) could underlie the differences between our results and those published previously (21).
Action-potential-evoked [Ca2+] transients could be modulated by presynaptic inhibitors. Adenosine has been suggested to act as a presynaptic inhibitor in a number of preparations (39–41). Previous imaging studies in brain slices have shown that adenosine attenuates calcium influx into populations of presynaptic terminals (35) (12). However, from these studies, it was not clear whether adenosine-dependent modulation was restricted to a subset of terminals or was expressed globally. We find that adenosine significantly attenuated the [Ca2+] transients in varicosities of cortical pyramidal cells. Importantly, adenosine had an effect on all terminals tested, suggesting that all terminals on layer 2/3 pyramidal cells share this mechanism of presynaptic inhibition. It will be of interest to test whether other modulators of presynaptic calcium influx are distributed homogeneously.
Acknowledgments
We thank Zach Mainen, Miguel Maravall, Bernardo Sabatini, and Barry Burbach for help with experiments and useful discussions. Support was provided by the National Institutes of Health (EY03038, to C.L.C.; 1R01NS38259-01, to K.S.) and the Klingenstein, Pew and Mathers Foundations (to K.S.).
Abbreviations
- VSCC
voltage-sensitive calcium channel
- [Ca2+]
intracellular free calcium concentration
- 2PLSM
two-photon excitation laser scanning microscopy
- ACSF
artificial cerebrospinal fluid
- OGB1
Oregon green 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA) I
Footnotes
See commentary on page 9349.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.170278697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.170278697
References
- 1.Braitenberg V, Schutz A. Anatomy of the Cortex: Studies of Brain Function. Vol. 18. Berlin: Springer; 1991. [Google Scholar]
- 2.Peters A, Palay S L, Webster HD. The Fine Structure of the Nervous System. New York: Oxford Univ. Press; 1991. [Google Scholar]
- 3.Mulkey R M, Zucker R S. Nature (London) 1991;350:153–155. doi: 10.1038/350153a0. [DOI] [PubMed] [Google Scholar]
- 4.Katz B. The Release of Neurotransmitter Substances. Springfield, IL: Thomas; 1969. [Google Scholar]
- 5.Wall P D. Trends Neurosci. 1995;18:99–103. doi: 10.1016/0166-2236(95)93883-y. [DOI] [PubMed] [Google Scholar]
- 6.Luscher H R, Shiner J S. Biophys J. 1990;58:1389–1399. doi: 10.1016/S0006-3495(90)82485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luscher C, Streit J, Lipp P, Luscher H R. J Neurophysiol. 1994;72:634–643. doi: 10.1152/jn.1994.72.2.634. [DOI] [PubMed] [Google Scholar]
- 8.Luscher C, Streit J, Quadroni R, Luscher H R. J Neurophysiol. 1994;72:622–633. doi: 10.1152/jn.1994.72.2.622. [DOI] [PubMed] [Google Scholar]
- 9.Sabatini B L, Regehr W G. Nature (London) 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- 10.Sabatini B L, Regehr W G. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regehr W G, Delaney K R, Tank D W. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L G, Saggau P. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 13.Llinas R, Sugimori M, Silver R B. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- 14.Helmchen F, Borst J G, Sakmann B. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callewaert G, Eilers J, Konnerth A. J Physiol (London) 1996;495:641–647. doi: 10.1113/jphysiol.1996.sp021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackenzie P J, Umemiya M, Murphy T H. Neuron. 1996;16:783–795. doi: 10.1016/s0896-6273(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie P J, Murphy T H. J Neurophysiol. 1998;80:2089–2101. doi: 10.1152/jn.1998.80.4.2089. [DOI] [PubMed] [Google Scholar]
- 18.Frenguelli B G, Malinow R. Learn Mem. 1996;3:150–159. doi: 10.1101/lm.3.2-3.150. [DOI] [PubMed] [Google Scholar]
- 19.Tan, Y. P. & Llano, I. (1999) J. Physiol. (London)520 Pt 1, 65–78. [DOI] [PMC free article] [PubMed]
- 20.Allen C, Stevens C F. Proc Natl Acad Sci USA. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debanne D, Guerineau N C, Gahwiler B H, Thompson S M. Nature (London) 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- 22.Denk W, Strickler J H, Webb W W. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 23.Denk W, Svoboda K. Neuron. 1997;18:351–357. doi: 10.1016/s0896-6273(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 24.Kincaid A E, Zheng T, Wilson C J. J Neurosci. 1998;18:4722–4731. doi: 10.1523/JNEUROSCI.18-12-04722.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shepherd G M, Harris K M. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mainen Z F, Malinow R, Svoboda K. Nature (London) 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- 27.Maravall M, Mainen Z M, Sabatini B, Svoboda K. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mainen Z F, Maletic-Savatic M, Shi S H, Hayashi Y, Malinow R, Svoboda K. Methods. 1999;18:231–239. doi: 10.1006/meth.1999.0776. [DOI] [PubMed] [Google Scholar]
- 29.Yuste R, Denk W. Nature (London) 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 30.Regehr W G, Atluri P P. Biophys J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debanne D, Kopysova I L, Bras H, Ferrand N. J Physiol (Paris) 1999;93:285–296. doi: 10.1016/s0928-4257(00)80057-1. [DOI] [PubMed] [Google Scholar]
- 32.Kopysova I L, Debanne D. J Neurosci. 1998;18:7436–7451. doi: 10.1523/JNEUROSCI.18-18-07436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke R E, Nerbonne J M. J Neurophysiol. 1997;78:2309–2320. doi: 10.1152/jn.1997.78.5.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gil Z, Connors B W, Amitai Y. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 35.Dittman J S, Regehr W G. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neher E, Augustine G J. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tank D W, Regehr W G, Delaney K R. J Neurosci. 1995;15:7940–7952. doi: 10.1523/JNEUROSCI.15-12-07940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helmchen F, Imoto K, Sakmann B. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunwiddie T V. Int Rev Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- 40.Greene R W, Haas H L. Prog Neurobiol. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- 41.Thompson S M, Capogna M, Scanziani M. Trends Neurosci. 1993;16:222–227. doi: 10.1016/0166-2236(93)90160-n. [DOI] [PubMed] [Google Scholar]