

Abstract
Reelin is a key mediator of ordered neuronal alignment in the brain. Here, we demonstrate that Reelin molecules assemble with each other to form a huge protein complex both in vitro and in vivo. The Reelin–Reelin interaction clearly is inhibited by the function-blocking anti-Reelin antibody, CR-50, at a concentration known to inhibit Reelin function. This assembly is mediated by electrostatic interaction of the CR-50 epitope region. Recombinant CR-50 epitope fragments spontaneously constitute a soluble, string-like homopolymer with a regularly repeated structure composed of more than 40 monomers. Mutated Reelin, which lacks the CR-50 epitope region, cannot form a homopolymer and fails to induce efficient tyrosine phosphorylation of Disabled 1 (Dab1), which should occur to transduce the Reelin signal. These data suggest that Reelin exerts its biological function by composing a large protein assembly driven by the CR-50 epitope region, proposing a novel model of the Reelin signaling in neurodevelopment.
In the mammalian central nervous system (CNS), various classes of neurons migrate from their site of origin to the final positions, where they are arranged in elaborate laminar structures (1–3). Neocortical development starts from the preplate formation. The preplate resides near the surface of the cortex and is composed of a superficial plexus of corticopetal nerve fibers and earliest-generated neurons, including the Cajal–Retzius and prospective subplate neurons. Consecutively, the preplate is split by the cortical plate neurons into a superficial marginal zone, where the Cajal–Retzius neurons differentiate, and a deep subplate, in which the subplate neurons differentiate (4). The cortical plate neurons are born in the ventricular zone and migrate past the intermediate zone and subplate along radial glial fibers before reaching the cortical plate. The systematic migration of the later-generated neurons past those generated earlier results in an “inside-out” progression in the mammalian cortical plate (5, 6).
The reeler is an autosomal recessive mouse mutant, in which neurons are generated normally but are abnormally placed, resulting in disorganization of cortical laminar layers in the CNS (7–12). In the reeler neocortex, for example, cortical plate neurons are aligned in a practically inverted fashion (“outside-in”). Therefore, the reeler mouse presents a good model in which to investigate the mechanisms of establishment of the precise neuronal network during development. We previously obtained a monoclonal alloantibody, CR-50, by immunizing reeler mice with homogenates of normal embryonic brains (13). This antibody was shown to react specifically with Cajal–Retzius neurons in the marginal zone of normal mice. It then was shown to recognize the Reelin protein itself (14), which is encoded by the reeler gene (reelin, Reln) (15, 16). Reelin is a secreted extracellular matrix protein composed of 3,461 aa with a relative molecular mass of 388 kDa (14, 15) (Fig. 1A). The N terminus of Reelin has 25% identity with that of F-spondin, an extracellular matrix protein that controls the adhesion and extension of commissural axons in the spinal cord (17). The first 500 aa of Reelin are followed by eight “Reelin repeats,” each of which is composed of 350–390 aa and contains an EGF-like motif of 30 aa in the middle (15). The highly charged C terminus of Reelin is essential for its secretion into the extracellular space (14, 18).
Figure 1.
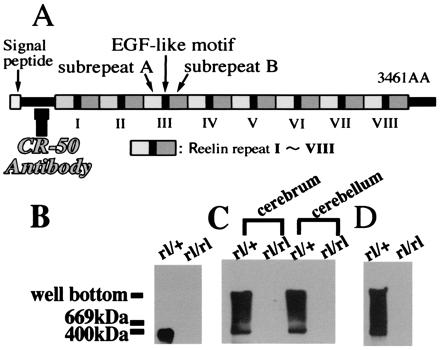
Native Reelin molecules form a large protein complex. (A) Schematic representation of the Reelin structure. CR-50 antibody recognizes upstream of the Reelin repeats. (B–D) Immunoblot with anti-Reelin antibody. (B) Homogenates of reeler heterozygote (rl/+) and homozygote (rl/rl) cerebra (E18) were run through SDS/PAGE. (C) Homogenates of cerebra (E18) and cerebella (P5) of reeler heterozygotes and homozygotes were separated by native gel. (D) Supernatants of the cerebellar cultures of reeler heterozygote and homozygote (P5) were loaded onto the native gels.
The CR-50 antibody has neutralizing activity of Reelin both in vitro and in vivo (13, 19–21). Recently, we demonstrated that the epitope of CR-50 is located near the N terminus (14) and since have defined it to amino acids 230–346 of Reelin (K.N., unpublished results). Because CR-50 inhibits the Reelin functions, this CR-50 epitope region should play a crucial role in the Reelin signaling during neurodevelopment.
In the cytoplasm, the Reelin signal is received by the cytosolic adapter protein, Disabled 1 (Dab1) (22–27), whose defect causes reeler-like phenotype in mice (23, 28, 29). Dab1 binds to nonphosphorylated NPXY motifs in the cytoplasmic domains of several cell surface proteins (30–33). Reelin induces tyrosine phosphorylation of Dab1 (24). It also was shown that the phenotype of double-knockout mice lacking both very low density lipoprotein receptor (VLDLR) gene and the apolipoprotein E receptor 2 (ApoER2) gene is indistinguishable from that of reeler mice (34). Recently, Reelin has been demonstrated to bind the VLDLR, the ApoER 2 (35, 36), and the cadherin-related neuronal receptor (CNR) family members (37), which then leads to Dab1 tyrosine phosphorylation. These results suggest that Reelin controls neuronal migration and positioning by stimulating tyrosine phosphorylation of Dab1 via VLDLR, ApoER2, and CNR family proteins.
In this study, we show that Reelin proteins bind to each other to form a large protein complex both in vitro and in vivo. Interestingly, this assembly formation clearly is inhibited by the CR-50 antibody at a concentration known to neutralize Reelin functions. We then show that the recombinant CR-50 epitope region indeed makes a regular homopolymer mediated by electrostatic interaction spontaneously. When the CR-50 epitope on Reelin was mutated, the recombinant protein no longer induced efficient tyrosine phosphorylation of Dab1 protein. These data suggest that Reelin exerts its biological function by composing a complex multimer through the CR-50 epitope region, proposing a novel model of the Reelin signaling in neurodevelopment.
Materials and Methods
Animals.
We used reeler mice originally derived from the B6C3Fe-a/a-rl adults (The Jackson Laboratory). The day at which a vaginal plug was detected and the day of birth were designated as E0 and P0, respectively.
Cerebeller Primary Culture.
P5 reeler cerebella were cultured as reported (38). The cultures were switched to serum-free medium 18 h after plating to be incubated for 4 days. CR-50 antibody was added to the serum-free medium from the beginning.
Western Blot Analysis.
Samples were run through SDS-containing gel or native gel, as described in the text, by using regular Laemmli buffer. Immunoblotting was performed with G10 anti-Reelin (kindly provided by A. Goffinet, University of Namur, Namur, Belgium), anti-FLAG (M2; Sigma), anti-phosphotyrosine (4G10; Upstate Biotechnology, Lake Placid, NY), or B3 anti-Dab1 antibody (kindly provided by J. Cooper and B. Howell, Fred Hutchinson Cancer Research Center, Seattle) followed by peroxidase-conjugate secondary antibody. The signal was visualized by chemiluminescent detection system (Boehringer Mannheim). One hundred micrograms of homogenate proteins or 20 μl of the culture supernatants was used.
Immunofluorescence.
293T cells were transfected with the pCrl full-length reelin construct (kindly provided by T. Curran, St. Jude Children's Research Hospital, Memphis, TN). Two days after transfection, cells were fixed and stained with CR-50 plus fluorescein isothiocyanate-conjugated secondary antibody as described before (13). In the case of living cell surface staining, CR-50 was added directly to the medium and incubated for 30 min at 37°C. Then, the cells were fixed and incubated with the secondary antibody.
Cell Adhesion Assay.
293T cells transfected with pCrl and those transfected with the vector pcDNA3 (Invitrogen) alone were cultured in DMEM + 10% FCS for 5 days, and the supernatants were collected. After nitrocellulose/methanol treatment, Petri dishes were coated with each supernatant or recombinant CR-50 epitope proteins for overnight. Then, 293T cells, which were transfected with pCrl or pcDNA3 2 days before, were plated onto the Reelin-coated or control-coated dishes. After a 2-h incubation, each plate was washed with panning buffer (PBS containing 10% FCS) until no floating cells remained. To examine the effect of CR-50 on cell adhesion, it was added to the culture medium from the beginning of incubation. Then, the cells remaining on precoated dishes were counted. This experiment was repeated five times, and for each dish, five microscopic fields were scored.
Expression and Purification of Reelin Epitope Protein.
The CR-50 epitope fragment (residues 230–346) was cloned into the pET-29a (Novagen), produced in BL21/pLysS Escherichia coli cells, and purified with a His⋅Bind resin affinity column (Novagen). It then was dialyzed against DTT-containing PBS (20 mM sodium phosphate, pH 7.4/0.15 M NaCl/5 mM DTT).
Gel Filtration Chromatography.
The FPLC gel filtration chromatography system (Pharmacia) with a Superdex-200 column (separation range: 10–600 kDa; Pharmacia) was used. The loaded sample was adjusted to 40 μM in DTT-containing PBS.
Electron Microscopy.
The whole images of CR-50 epitope polymer were observed by electron microscopy by using the rotary shadowing method and the negative staining method (39). The sample concentration was adjusted to 0.2 μM.
Congo Red Measurement.
Samples were tested for Congo red binding by the spectroscopic band-shift assay. Congo red analysis was performed as described previously (40–42).
Construction of the Mutated Reelin.
FLAG tag was put into the pSecTag2 vector (Invitrogen), and amino acids 368-3461 of Reelin were inserted downstream of the FLAG.
Results
Reelin Proteins Have Homophilic Interactions to Form a Large Protein Complex.
We showed previously that, when living embryonic brain cells are stained with anti-Reelin antibodies, the cell surface of Reelin-secreting cells, such as Cajal–Retzius neurons in the neocortex, are well labeled in punctate pattern (13, 18, 20, 21). These data suggest that Reelin proteins are assembled and anchored to the surface of a cell membrane of the Reelin-secreting cells. To determine whether the Reelin protein would form a large protein complex in vivo, we performed Western blot analysis after electrophoresis through both nondenaturing and denaturing gels. When embryonic cerebral homogenates [embryonic day 18 (E18)] were separated under the denaturing condition, immunoblotting with an anti-Reelin antibody revealed a band of 400 kDa in heterozygous reeler (Relnrl/+; rl/+) (Fig. 1B). This apparently corresponds to a monomer of the Reelin protein. In contrast, when cerebral (E18) and cerebellar [postnatal day 5 (P5)] homogenates were run through nondenaturing native gels, a broad range of bands spread from 400 kDa to the well were detected in addition to the monomer band in heterozygotes (rl/+) (Fig. 1C). We then investigated the status of Reelin secreted from cultured cerebellar granule cells. The supernatants of the primary cerebellar cultures of reeler homozygotes (rl/rl) and heterozygotes were resolved on nondenaturing native gel and probed with anti-Reelin antibody. Again, only the samples from heterozygous mice showed a wide spread of bands with molecular masses higher than 400 kDa (Fig. 1D). All these results taken together suggested that secreted Reelin is in a state of a large protein complex both in vitro and in vivo.
Therefore, we then examined whether Reelin indeed shows homophilic interaction in cell adhesion assays. Previously we demonstrated that some of the secreted Reelin is anchored to the surface of Reelin-producing brain cells (13, 18, 20, 21, 39). Thus, we stained living reelin-transfected 293T cells with CR-50 antibody to see whether the secreted Reelin is anchored to the 293T cells as well (Fig. 2A). Cell surface of the living 293T cells was stained in punctate pattern similarly to that of living brain cells, whereas fixed cells were stained diffusely in the cytoplasm (Fig. 2B). These results indicate that some of the secreted Reelin molecules are anchored to the surface of the transfected 293T cells. When reelin-transfected 293T cells were plated onto Reelin-coated dishes, much more cells bound to them (Fig. 2C) than to control dishes, which had been coated with supernatants containing no Reelin (Fig. 2 D and G). Control cells transfected only with a backbone vector did not bind to Reelin-coated or control dishes (Fig. 2 E– G). Thus, it is highly likely that Reelin molecules have a homophilic interaction. Indeed, when we previously screened an expression cDNA library with a fragment of Reelin to look for molecules interacting with it, we noticed that the population of reelin cDNA itself was increasingly enriched by the second and third screening (data not shown). Taken together, these results show that Reelin proteins bind to each other to form a large protein complex.
Figure 2.
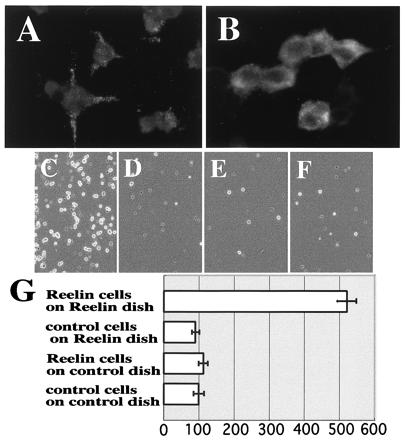
Cell adhesion assay indicates that Reelin molecules bind to each other. (A and B) Immunostaining of reelin-transfected 293T cells with CR-50. When CR-50 was added to the living cells, punctate staining on the cell surface was observed (A), whereas cytoplasm was stained diffusely when CR-50 was added after being fixed and permeablized (B). (C–F) Cell adhesion assay. (C) Reelin-presenting cells on Reelin-coated dish. (D) Reelin-presenting cells on control-coated dish. (E) Control cells on Reelin-coated dish. (F) Control cells on control-coated dish. (G) Histograms showing cell numbers that are normalized to F, which was scored as 100. Numbers are the mean value of five independent experiments ± SD.
Reelin Assembly Is Inhibited by the Function-Blocking CR-50 Antibody.
To investigate whether the assembly of Reelin is related to its physiological functions, we examined the effect of the function-blocking anti-Reelin antibody, CR-50 (13, 19–21), on this homophilic interaction. In the cell adhesion assay, when CR-50 antibody was added to the culture medium, it inhibited the adhesion in a dose-dependent manner (Fig. 3 A–E). At 20, 50, or 200 μg/ml CR-50 antibody, the number of bound cells was decreased to 56%, 44%, and 32%, respectively, of the control (Fig. 3A). Taking into account that the cell adhesion assay shows about 20% of nonspecific background binding (Fig. 2 D–G), 20, 50, and 200 μg/ml CR-50 antibody can inhibit the specific binding to approximately one-third, one-fourth, and one-tenth, respectively. Because the cell adhesion assay gives high background in general, because of technical difficulty, we further examined the effect of CR-50 antibody on the assembly of Reelin that is secreted into the medium. Cerebellar granule cell cultures were added with this antibody, and the supernatant was analyzed by Western blotting. Interestingly, even at 20 μg/ml CR-50 antibody, the broad high-molecular-weight bands were not detected at all (Fig. 3F). When nonimmunized mouse IgG was added to the culture medium, the homophilic Reelin complexes were formed normally. Thus, CR-50 antibody clearly inhibits the homophilic binding of Reelin even at 20 μg/ml. Because CR-50 antibody shows a function-blocking effect on Reelin from around this concentration (13), the inhibitory effect on homophilic interaction correlates well with the blocking activity of the Reelin function. These results suggest that the formation of homophilic Reelin complexes might have an essential role in its physiological function.
Figure 3.
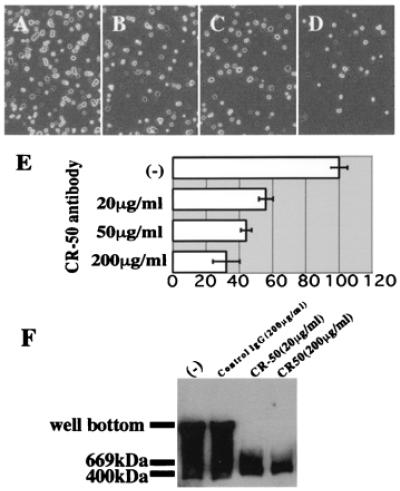
CR-50 antibody inhibits the homophilic interaction of Reelin. (A–E) Inhibition of cell adhesion by CR-50 antibody. (A) Reelin-presenting cells on Reelin-coated dish. (B–D) Reelin-presenting cells on Reelin-coated dish after incubation with 20, 50, and 200 μg/ml CR-50 antibody, respectively. (E) Histograms showing the cell numbers of each case (A–D) that are normalized to A. Numbers are the mean value of five independent experiments ± SD. (F) Inhibition of Reelin assembly by CR-50 antibody. reeler heterozygote cerebellar (P5) cells were cultured in the presence of no mouse IgG, nonimmunized mouse IgG, or CR-50 antibody. The supernatants of these cultures were separated by native gel and blotted with anti-Reelin.
To determine whether the Reelin assembly is mediated by the CR-50 epitope region itself, we coated Petri dishes with purified recombinant CR-50 epitope fragments and performed a cell adhesion assay by using full-length reelin-transfected 293T cells. When Reelin-presenting cells were plated onto the CR-50 epitope-coated dishes, much more cells bound to them in a dose-dependent manner than to the control dish (Fig. 4). These results indicate that the Reelin protein binds to the purified CR-50 epitope fragments, suggesting that the homophilic interaction is mediated by the CR-50 epitope region itself.
Figure 4.
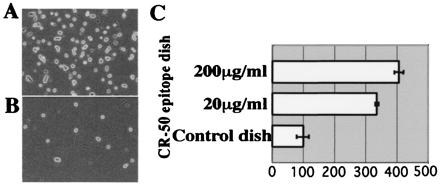
Reelin binds to the CR-50 epitope region in the cell adhesion assay. (A) Reelin-presenting cells on CR-50 epitope-coated dish (200 μg/ml). (B) Reelin-presenting cells on control-coated dish. (C) Histograms showing the cell numbers in the adhesion assay on CR-50 epitope-coated (200 or 50 μg/ml) dishes that are normalized to the control.
CR-50 Epitope Fragments Form Soluble Homopolymers.
The above-mentioned experiments showed that the CR-50 epitope on Reelin is a critical region in assembly formation. Thus, we then investigated the structural and physical features of the CR-50 epitope. We first analyzed the CR-50 epitope fragments by analytical gel filtration chromatography to see whether the epitope could assemble to form a homopolymer (Fig. 5). Under physiological conditions (0.15 M NaCl, pH 7.4), the elution profile of the CR-50 epitope fragments showed only a polymer peak at the void volume (V0) of this column (separation range: 10–600 kDa) (Fig. 5A). This suggests that all of the fragments were associated in a huge complex with a molecular mass larger than 600 kDa. The molecular mass of the CR-50 epitope monomer is about 15 kDa, so this polymer is composed of more than 40 CR-50 epitope molecules. Even in the presence of 10 mM DTT, this polymer was detected. To study the mechanism of polymer formation, we examined the effect of ionic strength on polymerization. As salt (NaCl) concentration increased, the polymer was gradually dissociated to monomers, indicating that this polymer is formed by electrostatic interaction (Fig. 5). Finally, at 1.0 M NaCl, all fragments were present as monomers (Fig. 5D). This transition from polymer to monomer was reversible depending on the salt concentration.
Figure 5.
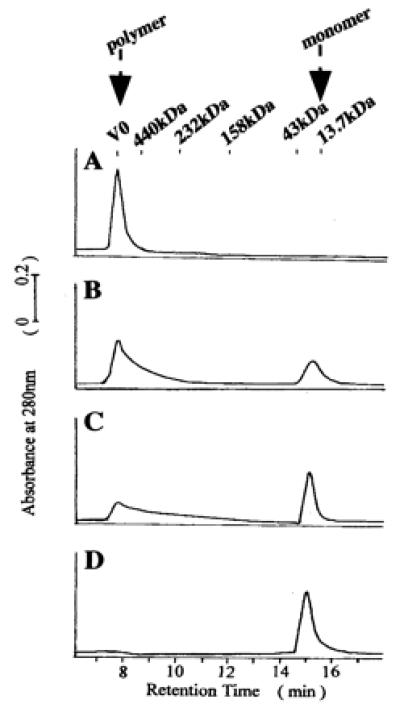
CR-50 epitope fragments form a homopolymer. (A) The elution profile of the gel filtration chromatography shows that CR-50 epitope fragments form a homopolymer with a molecular mass of more than 600 kDa at 0.15 M NaCl (A). The polymer is dissociated at a higher ionic strength [0.5 M NaCl (B); 0.75 M NaCl (C); 1.0 M NaCl (D)]. The polymer peak appears at an elution time of 7.7 min and a monomer peak at 15.2 min. V0 indicates the void volume of column.
The CR-50 epitope solution was transparent without any insoluble sediments, which often are experienced by irregular aggregation when using bacterially produced recombinant proteins. To confirm further that the polymer is different from the irregular aggregates, we performed Congo red-binding assay (40–42). Congo red is known to bind amyloid-like fibrils with a β-sheet structure in a regular parallel arrangement, which can be detected by a red-shift in the Congo red absorbance spectrum (42). Addition of the CR-50 epitope polymer to Congo red solution induced the red-shift in the λmax from 486 nm [control: shown as (b) in Fig. 6A] to 505 nm [Fig. 6A, (a)], which should not occur with irregular aggregates. The point of maximal spectral difference between the polymer-containing solution and dye-only solution was at 528 nm (Fig. 6B), which is shorter than that of amyloid fibrils (541 nm; ref. 42). Therefore, CR-50 epitope fragments formed a linear, soluble polymer with a regular structure, but that was different than amyloid fibrils.
Figure 6.
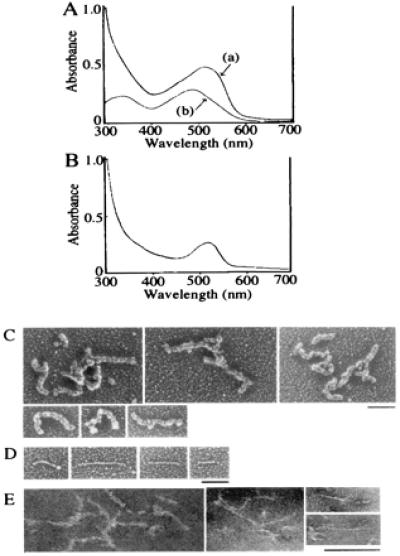
Structural analysis of CR-50 epitope polymer. (A) The absorbance spectra of Congo red with CR-50 epitope polymer (a) and Congo red alone (b) were observed. The red-shift of λmax occurred when CR-50 epitope polymer was added. (B) The spectral difference between the polymer-containing solution and dye-only solution was detected. (C–E) Electron micrographs of CR-50 epitope polymer by rotary shadowing method (C and D) or negative staining method (E). (Bar = 100 nm.)
We then investigated the structure of the CR-50 epitope polymer by using electron microscopy. Electron micrographs revealed numerous, string-like polymer structures, 100–400 nm in length regardless of the presence of 10 mM DTT (Fig. 6 C–E). The polymers shown in Fig. 6C appear to have a twisted structure of finer polymers. Some (less than 3% of the whole population) finer polymers with straight structure also were observed (Fig. 6D).
Mutated Reelin That Cannot Form a Homopolymer Fails to Induce Efficient Tyrosine Phosphorylation of Dab1.
Finally, we investigated whether the assembly formation related to the Reelin function. Mutant Reelin, in which the N terminus including the CR-50 epitope region was replaced with a FLAG tag, did not show assembly on Western blot analysis (Fig. 7A), further supporting the importance of the CR-50 epitope region in the assembly formation. In the cell adhesion assay, mutant reelin-transfected 293T cells did not bind to intact Reelin-coated dishes. Intact reelin-transfected 293T cells also did not bind to mutant Reelin-coated dishes (Fig. 7B). From these results, it was confirmed that the mutant Reelin lacking the CR-50 epitope could not assemble.
Figure 7.

Mutated Reelin that cannot form a homopolymer fails to induce efficient tyrosine phosphorylation of Dab1. (A) Western blot of mutated Reelin, which lacks the CR-50 epitope region, indicates that it cannot assemble to form a polymer. (B) Cell adhesion assay using mutated Reelin. Histograms show the cell numbers that are normalized to the control. (C) Cortical neurons obtained from E14 rl/rl mice were cultured and treated with supernatants of intact reelin-transfected cells, mutated reelin-transfected cells, or vector-transfected cells at 37°C for 30 min. Cells were solubilized and subjected to Western blot analysis. Although intact Reelin can efficiently induce tyrosine phosphorylation of Dab1, the mutated Reelin cannot. An applied amount of Dab1 protein was visualized by immunoblot analysis with anti-Dab1 antibody.
We then investigated the capability of the mutated Reelin to transduce signals. Tyrosine phosphorylation of Dab1 induced by mutated Reelin was reduced dramatically compared with the intact Reelin (Fig. 7C). These results highly suggest that assembly formation indeed is essential in efficient Reelin signaling.
Discussion
The data presented here indicate that Reelin molecules assemble together to form a huge protein complex both in vitro and in vivo. As we showed previously (13, 18, 20, 21) and in this study (Fig. 2A), some of the secreted Reelin is anchored to the surface of the Reelin-producing cells by an unknown mechanism. When Reelin proteins on living cells are immunostained with antibodies, they are labeled as many punctates on the cell membrane, which may reflect the matrix of Reelin assembly. In addition, we have shown here that the Reelin proteins secreted from cultured cells also form the homophilic complex in the medium. Thus, Reelin is thought to assemble irrespective of whether it is anchored to the cell surface or secreted into the extracellular space. When the Reelin receptor-expressing cells migrate and interact with Reelin, the homophilic Reelin complex may function as “multivalent ligands” and induce receptor polymerization, resulting in transphosphorylation of the intracellular Dab1 proteins by recruiting protein kinases. This receptor polymerization may have been observed as “dots” that are colocalized with Reelin when embryonic brains were double-stained with anti-CNR protein and CR-50 antibodies (37).
The assembly formation of Reelin is thought to be accomplished by electrostatic interaction via the CR-50 epitope region, which is localized near the N terminus of Reelin. CR-50 antibody appears to recognize a three-dimensional structure because it is very sensitive to various treatments such as fixation and ethanol treatment, and the epitope recently has been mapped to a region of 116 aa (K.N., unpublished results). This epitope region is essential for the Reelin functions, because CR-50 antibody inhibits laminar pattern formation in cortical reaggregation cultures (13), Purkinje cell alignment in cerebellar explant cultures (20), and an axonal branching pattern in the entorhinohippocampal pathway in vitro (19) and disrupts hippocampal development in vivo (21). The results of the present study indicate that the assembly of secreted Reelin is inhibited by CR-50 antibody even at 20 μg/ml. Because Reelin function is inhibited by the CR-50 antibody from around this concentration, it is highly likely that the Reelin assembly is perturbed when its function is blocked.
Recently, lipoprotein receptors (VLDLR and ApoER2) and the CNR family members were reported to bind to the full-length Reelin (35–37). However, VLDLR and ApoER2 do not bind to a recombinant N-terminal portion of Reelin, which includes the CR-50 epitope region (36), and the CNR proteins bind to the subrepeat B of the first Reelin repeat, which is downstream of the CR-50 epitope region (37). These results indicate that the binding sites of all three are located outside of the CR-50 epitope region on Reelin. Further analysis, however, revealed that CR-50 antibody inhibited Reelin binding to VLDLR-expressing 293T cells (35) and to the extracellular domain of CNR1 protein (37). One possible interpretation of these results is that CR-50 binding to the N terminus might inhibit the receptor–Reelin interaction indirectly by inducing structural change or steric hindrance to the binding sites on Reelin for all of these receptors. Alternatively, or additionally, CR-50 antibody may have decreased the number of Reelin proteins bound to the receptors by interfering with the huge homophilic complex formation of the ligand (Reelin) itself. Because the assembly of Reelin is inhibited by the CR-50 antibody, each receptor may have bound to a monomer rather than to a huge complex of Reelin, which should have been difficult to be distinguished from the direct inhibition of the receptor–ligand interaction. Interestingly, the assembly of Reelin that was secreted from cerebellar granule cells was inhibited even at 20 μg/ml antibody (Fig. 3F). In contrast, Reelin binding to the lipoprotein receptor-expressing cells was reduced to 36% and 16% at 115 and 300 μg/ml CR-50 antibody, respectively (35). The cell adhesion assay in the present study using full-length Reelin showed a practically similar level of inhibition by this antibody (Fig. 3E; reduced to 44% and 32% at 50 and 200 μg/ml CR-50 antibody). Taking into account that the adhesion assay using cultured cells gives high background in general (Figs. 2 D–G and 4 B and C), these data collectively suggest that the CR-50 antibody might neutralize Reelin function (at least in part) by inhibiting the homophilic assembly formation of the Reelin protein itself. It is thus important to clarify whether the binding of a Reelin monomer to the receptors could initiate appropriate signaling to control neuronal migration. The mutated Reelin, in which the CR-50 epitope region was deleted, did not form a polymer and was unable to induce efficient tyrosine phosphorylation of Dab1 (Fig. 7C). Because the Dab1 phosphorylation is essential in Reelin signaling (24, 35–37), these data implicate that Reelin exerts its biological functions by forming a homopolymer, which is inhibited by the CR-50 antibody.
Purified CR-50 epitope fragments are assembled spontaneously to form a soluble homopolymer via electrostatic interaction. Circular dichroism spectroscopy indicates that it has an α-helix (20%) and β-sheet (40%) structure, of which the α-helix region contributes mainly to the polymer formation (N.U.-T., K.N., S.T., M.K., and K.M., unpublished results). The secondary structure prediction by the method of Chou and Fasman (43) suggested that the epitope should have a long α-helix domain in its C terminus and a short α-helix domain in the middle. In fact, these domains contain many (≈30%) charged amino acids, which appear to be responsible for the polymerization. One possibility is that these two domains function as hinges to assemble monomers to make the linear, string-like structure of the polymer. One interesting finding in the gel filtration analysis was that under physiological conditions there was only a huge polymer peak, but no monomer peak (Fig. 5A). However, Western blot analysis of the full-length Reelin showed a broad range of bands from the monomer to the top of a native gel (Fig. 1 B–D), which indicates that there are various polymers composed of different numbers of monomers. This difference suggests that the CR-50 epitope region itself should assemble spontaneously, which then might drive full-length polymer formation. In the case of the full-length molecule, steric hindrance or some other mechanisms might keep them at various stages of polymerization.
Recently, we found a truncated isoform of reelin in Xenopus, which contains the CR-50 epitope region but none of the repeat region (H. Tabata, K.N., and K.M., unpublished results). This novel isoform should not bind to the lipoprotein receptors or the CNR proteins, because it has only the Reelin N terminus, but appears to be involved in the CR-50 epitope-mediated assembly formation. It would be interesting to determine whether the full-length Reelin multimer formation is regulated by competition with the truncated isoform. In any event, the mechanism of Reelin signaling might be much more complicated than ever thought.
In conclusion, our results indicate that the Reelin proteins interact with each other to form a huge, homophilic protein complex in the developing brain. This assembly is mediated by the epitope region against its function-blocking antibody, CR-50, which clearly disrupts this interaction. Mutated Reelin, which lacks the CR-50 epitope region, cannot assemble and fails to induce efficient tyrosine phosphorylation of Dab1. These results suggest that Reelin functions as a large, homophilic complex to control neuronal migration in neurodevelopment.
Acknowledgments
We appreciate Dr. M. Ogawa for his continuous support and valuable comments on the manuscripts. We also thank Dr. A. M. Goffinet for the G10 antibody, Dr. T. Curran for the pCrl plasmid, Drs. J. Cooper and B. Howell for the B3 anti-Dab1 antibody, and Dr. T. Ohshima for critical comments on the manuscript. The reeler mice were cleaned by in vitro fertilization and embryo transfer by the Division of Experimental Animal Research of RIKEN. This research was supported by grants from the ministry of Education, Science, Sports, and Culture of Japan, RIKEN, the Human Frontier Science Program, PRESTO [Japan Science and Technology Corporation (JST)], CREST (JST), The Sumitomo Foundation, Uehara Memorial Foundation, and NOVARTIS Foundation (Japan) for the Promotion of Science. K.K. is a junior research fellow of the Japan Society for the Promotion of Science.
Abbreviations
- Dab1
Disabled 1
- VLDLR
very low density lipoprotein receptor
- CNR
cadherin-related neuronal receptor
- En
embryonic day n
- ApoER2
apolipoprotein E receptor 2
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160272497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160272497
References
- 1.Rakic P. Proc Natl Acad Sci USA. 1995;92:11323–11327. doi: 10.1073/pnas.92.25.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearlman A L, Faust P L, Hatten M E, Brunstrom J E. Curr Opin Neurobiol. 1998;8:45–54. doi: 10.1016/s0959-4388(98)80007-x. [DOI] [PubMed] [Google Scholar]
- 3.Rice D S, Curran T. Genes Dev. 1999;13:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- 4.Allendoerfer K L, Shatz C J. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- 5.Angevine J B, Sidman R L. Nature (London) 1961;25:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 6.Rakic P. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 7.Falconer D S. J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 8.Rakic P, Caviness V S J. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- 9.Stanfield B B, Cowan W M. J Comp Neurol. 1979;185:423–459. doi: 10.1002/cne.901850303. [DOI] [PubMed] [Google Scholar]
- 10.Caviness V S., Jr Dev Brain Res. 1982;4:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 11.Caviness V S, Jr, Sidman R L. J Comp Neurol. 1973;148:141–151. doi: 10.1002/cne.901480202. [DOI] [PubMed] [Google Scholar]
- 12.de Rouvroit C L, Goffinet A M. Adv Anat Embryol Cell Biol. 1998;150:1–106. [PubMed] [Google Scholar]
- 13.Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Yamamoto H, Mikoshiba K. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 14.D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Arcangelo G, Miao G G, Chen S-C, Soares H D, Morgan J I, Curran T. Nature (London) 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 16.Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S, et al. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- 17.Klar A, Baldassare M, Jessell T M. Cell. 1992;69:95–110. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- 18.de Berbeyck V, Nakajima K, de Rouvroit C L, Naerhuyzen B, Goffinet A M, Miyata T, Ogawa M, Mikoshiba K. Mol Brain Res. 1997;50:85–90. doi: 10.1016/s0169-328x(97)00166-6. [DOI] [PubMed] [Google Scholar]
- 19.Del Rio J Q, Heimich B, Borrell V, Forster E, Drakew A, Alcantara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, et al. Nature (London) 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- 20.Miyata T, Nakajima K, Mikoshiba K, Ogawa M. J Neurosci. 1997;17:3599–3609. doi: 10.1523/JNEUROSCI.17-10-03599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Proc Natl Acad Sci USA. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howell B W, Gertler F B, Cooper J A. EMBO J. 1997;16:121–132. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howell B W, Hawkes R, Soriano P, Cooper J A. Nature (London) 1997b;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 24.Howell B W, Herrick T M, Cooper J A. Genes Dev. 1999;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheldon M, Rice D S, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell B W, Cooper J A, Goldowitz D, Curran T. Nature (London) 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 26.Ware M L, Fox J W, Gonzales J L, Davis N M, de Rouvroit C L, Russo C J, Chua S C, Jr, Goffinet A M, Walsh C A. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- 27.Rice D S, Sheldon G, D'Arcangelo G, Nakajima K, Goldowitz D, Curran T. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- 28.Sweet H O, Bronson R T, Johnson K R, Cook S A, Davisson M T. Mamm Genome. 1996;7:798–802. doi: 10.1007/s003359900240. [DOI] [PubMed] [Google Scholar]
- 29.Yoneshima H, Nagata E, Matsumoto M, Yamada M, Nakajima K, Miyata T, Ogawa M, Mikoshiba K. Neurosci Res. 1997;29:217–223. doi: 10.1016/s0168-0102(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 30.Pawson T, Scott J D. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 31.Trommsdorff M, Borg J P, Margolis B, Herz J. J Biol Chem. 1998;273:3556–3560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 32.Homayouni R, Rice D S, Sheldon M, Curran T. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howell B, Lanier L M, Frank R, Gertler F B, Cooper J A. Mol Cell Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimph J, Hammer R E, Richerdson J A, Herz J. Cell. 1999;97:698–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 35.D'Arcangelo G, Homayouni R, Keshvara L, Rice D S, Sheldon M, Curran T. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 36.Hiesberger T, Trommsdorff M, Howell B, Goffinet A, Mumby M C, Cooper J A, Herz J. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 37.Senzaki K, Ogawa M, Yagi T. Cell. 1999;99:635–647. doi: 10.1016/s0092-8674(00)81552-4. [DOI] [PubMed] [Google Scholar]
- 38.Miyata T, Nakajima K, Aruga J, Takahashi S, Ikenaka K, Mikoshiba K, Ogawa M. J Comp Neurol. 1996;372:215–228. doi: 10.1002/(SICI)1096-9861(19960819)372:2<215::AID-CNE5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 39.Katayama E. J Biochem. 1989;106:751–70. doi: 10.1093/oxfordjournals.jbchem.a122928. [DOI] [PubMed] [Google Scholar]
- 40.Klunk W E, Pettegrew J W, Abraham D J. J Histochem Cytochem. 1989a;37:1273–1279. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 41.Klunk W E, Pettegerw J W, Abraham D J. J Histochem Cytochem. 1989b;37:1293–1297. doi: 10.1177/37.8.2666512. [DOI] [PubMed] [Google Scholar]
- 42.Klunk W E, Jacob R F, Mason R P. Anal Biochem. 1999;266:66–76. doi: 10.1006/abio.1998.2933. [DOI] [PubMed] [Google Scholar]
- 43.Chou P Y, Fasman G D. Adv Enzymol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]